





توضیحات محصول
توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
توالی یابی کل ژنوم
توالییابی کل ژنوم (WGS)، که بهعنوان توالییابی ژنوم کامل یا توالییابی کل ژنوم نیز شناخته میشود، فرآیند تعیین کل یا تقریباً کل توالی DNA ژنوم یک موجود زنده در یک زمان واحد است. این امر مستلزم تعیین توالی تمام DNA کروموزومی یک موجود زنده و همچنین DNA موجود در میتوکندری و برای گیاهان توالی موجود در کلروپلاست میباشد.
توالی یابی کل ژنوم تا حد زیادی به عنوان یک ابزار تحقیقاتی مورد استفاده قرار گرفته است، اما در سال 2014 به کلینیک ها معرفی شد. در آینده پزشکی شخصی، دادههای توالی ژنوم ممکن است ابزار مهمی برای هدایت مداخلات درمانی باشد. ابزار تعیین توالی ژن در سطح SNP نیز برای مشخص کردن انواع عملکردی از مطالعات ارتباطی و بهبود دانش در دسترس محققان علاقهمند به زیستشناسی تکاملی استفاده میشود و از این رو ممکن است پایهای برای پیشبینی حساسیت به بیماری و پاسخ دارویی ایجاد کند.
توالی یابی کل ژنوم نباید با پروفایل DNA اشتباه گرفته شود. پروفایل DNA فقط احتمال اینکه مواد ژنتیکی از یک فرد یا گروه خاص آمده باشد را تعیین می کند و حاوی اطلاعات اضافی در مورد روابط ژنتیکی، منشاء یا استعداد ابتلا به بیماری های خاص نیست. به علاوه توالی یابی کل ژنوم را نباید با روش هایی اشتباه گرفت که زیرمجموعه های خاص ژنوم را توالی می کند – چنین روش هایی شامل توالی یابی کل اگزوم (1-2٪ از ژنوم) یا SNP ژنوتیپ (0.1%> از ژنوم) است.
سلول های مورد استفاده برای توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
تقریباً هر نمونه بیولوژیکی حاوی یک کپی کامل ازDNA (حتی مقدار بسیار کمی از DNA یا DNA قدیمی) میباشد که میتواند مواد ژنتیکی لازم برای توالی ژنوم کامل را فراهم کند. چنین نمونه هایی ممکن است شامل بزاق، سلول های اپیتلیال، مغز استخوان، مو (تا زمانی که مو دارای فولیکول مو باشد)، دانه ها، برگ های گیاه یا هر چیز دیگری که دارای سلول های حاوی DNA است، باشد.
توالی ژنوم یک سلول منفرد انتخاب شده از یک جمعیت مخلوط سلولی را می توان با استفاده از تکنیک های توالی یابی ژنوم تک سلولی تعیین کرد. این موارد از مزیتهای مهمی در میکروبیولوژی محیطی به شمار میرود در مواردی به این صورت که یک سلول منفرد از یک گونه میکروارگانیسم خاص را می توان بر اساس ویژگی های مورفولوژیکی یا سایر ویژگی های متمایز کننده از یک جمعیت مخلوط با میکروسکوپ جدا کرد.
توالی ژنوم تک سلولی به عنوان روشی برای تشخیص ژنتیکی قبل از لانه گزینی در حال آزمایش است، که در آن یک سلول از جنین ایجاد شده توسط لقاح آزمایشگاهی گرفته شده و قبل از انتقال جنین به رحم تجزیه و تحلیل می شود. پس از کاشت، با آزمایشcell-free میتوان DNA جنین رابا رگ گیری ساده از مادر گرفته و برای تعیین توالی ژنوم کل جنین استفاده کرد.
تکنیک های اولیه توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
توالی یابی تقریباً کل ژنوم انسان برای اولین بار در سال 2000 تا حدی با استفاده از فناوری توالی یابی تفنگ ساچمه ای انجام شد. با شروع پروژه های توالی یابی ژنوم های طولانی تر و پیچیده تر، گروه های متعدد متوجه شدند که اطلاعات مفیدی را می توان با تعیین توالی هر دو انتهای یک قطعه DNA به دست آورد. اگرچه تعیین توالی هر دو انتهای یک قطعه و ردیابی داده های جفت شده دشوارتر از توالی یابی یک انتهای واحد از دو قطعه متمایز بود، اما دانستن اینکه دو توالی در جهت مخالف قرار گرفته اند و تقریباً طول یک قطعه جدا از هر یک می باشد، در بازسازی توالی هدف اصلی ارزشمند بود.
اولین توصیف منتشر شده در مورد استفاده از انتهای جفت شده در سال 1990 به عنوان بخشی از توالی یابی لوکوسHPRT انسانی بود. اولین تئوری از استراتژی توالی یابی انتهایی دوتایی خالص، با فرض قطعاتی با طول ثابت، در سال 1991 بود. در سال 1995، نوآوری استفاده از قطعات با اندازه های مختلف معرفی شد، و نشان داد که استراتژی توالی انتهایی مزدوج خالص در اهداف بزرگ امکان پذیر است. این استراتژی متعاقباً توسط مؤسسه تحقیقات ژنومی TIGR برای تعیین توالی کل ژنوم باکتری هموفیلوس آنفولانزا در سال 1995، و سپس توسط Celera Genomics برای تعیین توالی کل ژنوم مگس میوه در سال 2000، و به دنبال آن توالی یابی کل ژنوم انسان انجام شد. Applied Biosystems که اکنون Life Technologies نامیده می شود، توالی سنجی مویرگی خودکار را که توسط Celera Genomics و پروژه ژنوم انسانی استفاده می شود را تولید کرد.
تکنیک های فعلی
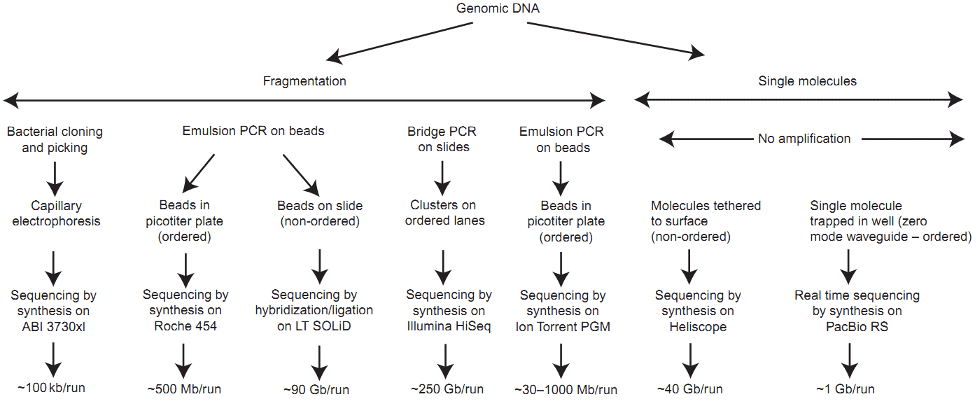
در حالی که توالی یابی مویرگی اولین رویکرد برای توالی یابی موفقیت آمیز ژنوم تقریباً کامل انسان بود، هنوز هم بسیار گران است و برای اهداف تجاری بسیار طولانی است. از سال 2005 توالی یابی مویرگی به تدریج توسط فناوری های توالی یابی با توان بالا (نسل بعدی) مانند توالی یابی رنگ ایلومینا، پایروسکوئنسینگ و توالی یابی SMRT جابجا شده است. همه این فناوریها از استراتژی اصلی تفنگ ساچمهای، یعنی موازیسازی و تولید الگو از طریق قطعهسازی ژنوم بهره میبرند.
فناوریهای دیگری از جمله فناوری Nanoporeظهور کردهاند. اگرچه دقت توالی فناوری Nanopore کمتر از موارد بالا است، اما طول خواندن آن به طور متوسط بسیار طولانیتر است. این نسل از خواندن طولانی به ویژه در کاربردهای جدید توالی یابی کل ژنوم ارزشمند است.
تحلیل و بررسی توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
در اصل، توالی یابی کامل ژنوم می تواند توالی نوکلئوتیدی خام DNA یک ارگانیسم در یک نقطه از زمان را فراهم کند. با این حال، تجزیه و تحلیل بیشتر باید انجام شود تا معنای بیولوژیکی یا پزشکی این توالی ارائه شود، مانند اینکه چگونه می توان از این دانش برای کمک به پیشگیری از بیماری استفاده کرد. روش هایی برای تجزیه و تحلیل داده های توالی یابی در حال توسعه و اصلاح هستند.
از آنجایی که توالییابی، دادههای زیادی تولید میکند (به عنوان مثال، تقریباً شش میلیارد جفت باز در هر ژنوم دیپلوئید انسانی وجود دارد)، خروجی آن به صورت الکترونیکی ذخیره میشود و به مقدار زیادی توان محاسباتی و ظرفیت ذخیرهسازی نیاز دارد.
در حالی که تجزیه و تحلیل داده های WGS می تواند کند باشد، می توان با استفاده از سخت افزار اختصاصی این مرحله را سرعت بخشید.
دانشمندان دریافته اند که توالی یابی کل ژنوم به طور قوی شایع ترین اختلالات عصبی ارثی را تشخیص می دهد، چیزی که قبلا تصور می شد غیرممکن است.
توالی یابی کل ژنوم توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing چگونه کار می کند؟
دانشمندان با دنبال کردن این چهار مرحله اصلی، توالی یابی کل ژنوم را انجام می دهند:
برش DNA: دانشمندان با استفاده از قیچی مولکولی DNA را که از میلیونها باز (A، C، T وG) تشکیل شده است، به قطعاتی میبرند که به اندازه کافی کوچک هستند تا دستگاه توالییابی بتواند آن را بخواند.
نوار کدگذاری DNA: دانشمندان تکه های کوچکی از برچسب های DNA یا کدهای نواری را اضافه می کنند تا مشخص کنند کدام قطعه DNA بریده شده متعلق به کدام باکتری است. این شبیه به نحوه شناسایی یک بارکد یک محصول در یک فروشگاه مواد غذایی است.
تعیین توالی DNA: DNA بارکد شده از چندین باکتری ترکیب شده و در یک توالی سنجی DNA قرار می گیرد. توالیسنج A، C، T و G یا پایههایی را که هر توالی باکتری را تشکیل میدهند، شناسایی میکند. توالیسنج از بارکد استفاده میکند تا ردیابی پایگاههایی را که به کدام باکتری تعلق دارند، ردیابی کند.
تجزیه و تحلیل داده ها: دانشمندان از ابزارهای تحلیل کامپیوتری برای مقایسه توالی از چندین باکتری و شناسایی تفاوت ها استفاده می کنند. تعداد تفاوتها میتواند به دانشمندان بگوید که این باکتریها چقدر به هم نزدیک هستند و چقدر احتمال دارد که بخشی از یک شیوع باشند.
توالی یابی کل ژنوم بزرگ
تعیین توالی ژنوم های بزرگ (> 5 مگابایت)، مانند ژنوم انسان، گیاه یا حیوان، می تواند اطلاعات ارزشمندی را برای تحقیقات بیماری و ژنتیک جمعیت ارائه دهد.
توالی یابی کل ژنوم کوچک
توالی یابی ژنوم کوچک (≤ 5 Mb) شامل تعیین توالی کل ژنوم یک باکتری، ویروس یا میکروب دیگر است. بدون نیاز به کشت باکتریایی، محققان می توانند هزاران ارگانیسم کوچک را به صورت موازی با استفاده از NGS توالی یابی کنند.
توالی یابی De Novo
توالی یابی De novo به تعیین توالی یک ژنوم جدید اشاره دارد که در آن توالی مرجعی در دسترس نباشد. NGS امکان شناسایی سریع و دقیق هر گونه را فراهم می کند.
توالی یابی مرحله ای
توالی یابی فازی یا فازبندی ژنوم، بین آلل های کروموزوم های همولوگ تمایز قائل می شود که منجر به هاپلوتیپ های کل ژنوم می شود. این اطلاعات اغلب برای مطالعات بیماری های ژنتیکی مهم است.
توالی یابی کل ژنوم انسان
قبلاً یک برنامه چالش برانگیزبوده، توالی یابی کل ژنوم انسان هرگز به سادگی که امروزه صورت میگیرد نبوده است. این جزئیات دقیقی را در کد ژنتیکی ما ارائه می دهد.
نحوه انجام WGS
دو رویکرد کلاسیک برای تعیین توالی ژنوم های بزرگ توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
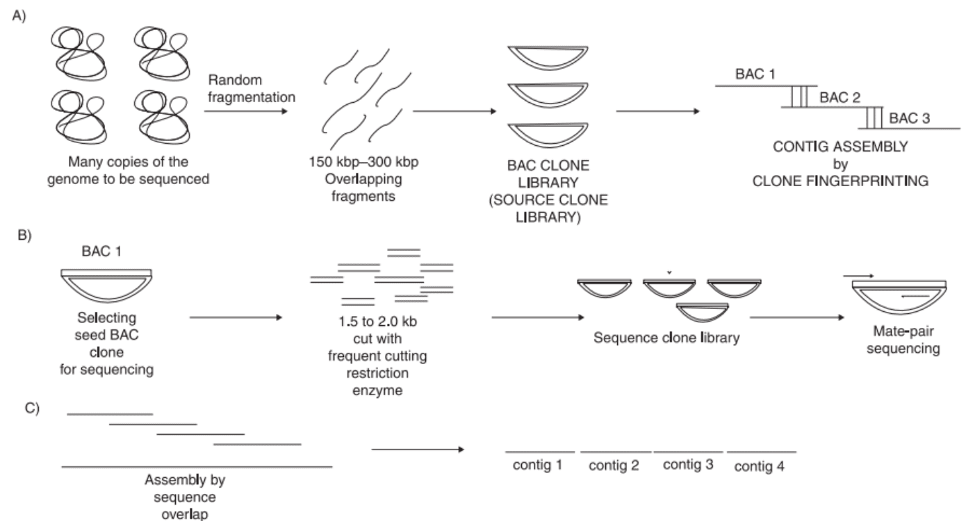
در اوایل دهه 80، سانگر با استفاده از روش تفنگ ساچمه ای، توالی یابی کل ژنوم فاژ لامبدا را با موفقیت انجام داد و این روش با موفقیت در DNA ویروس بزرگتر، DNA اندامک و تعیین توالی DNA ژنوم باکتری به کار رفت. توالی یابی شاتگان یک استراتژی کلاسیک برای توالی یابی کل ژنوم است. استراتژی توالی تفنگ ساچمه ای تضمینی فنی برای توالی یابی در مقیاس بزرگ فراهم می کند. این فناوری ابتدا به طور تصادفی یک توالی هدف کامل را به قطعات کوچک قطع میکند و به صورت جداگانه توالی مییابد، و سپس با استفاده از روابط همپوشانی این قطعات کوچک، آنها را به دنبالهای ثابت تقسیم میکند. این عمدتا شامل دو روش است: یکی توالی یابی سلسله مراتبی تفنگ ساچمه ای (روش کلون به کلون) و دیگری توالی یابی تفنگ ساچمه ای کل ژنوم است.
- توالی یابی کلون به کلون
این روش زمانی توسط کنسرسیوم HGP اتخاذ شد. این روش می تواند نقشه هایی با چگالی بالا ایجاد کند و مونتاژ ژنوم را آسان تر کند. این به طور کلی شامل چهار مرحله، آماده سازی کتابخانه کلون BAC، آماده سازی اثر انگشت کلون، توالی یابی کلون BAC و مونتاژ توالی است. با این حال، این روش زمان بر و پرهزینه است، بنابراین در حال حاضر به ندرت از آن استفاده می شود.
whole genome sequencing
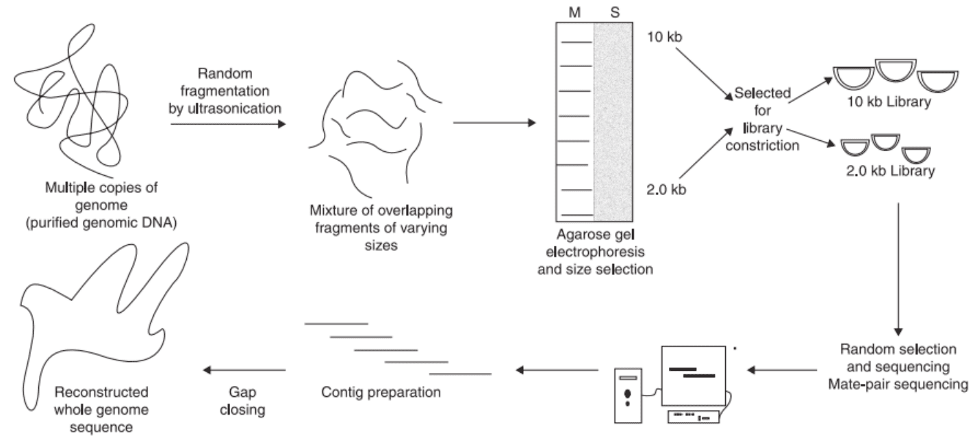
- توالی تفنگ ساچمهای کل ژنوم (WGS)
WGS به طور کلی شامل شش مرحله، جداسازی DNA ژنومی، تکه تکه شدن تصادفی DNA ژنومی، انتخاب اندازه با استفاده از الکتروفورز، ساخت کتابخانه، توالی یابی زوجی و مونتاژ ژنوم است. دو اندازه مختلف از قطعات DNA شامل قطعه الحاقی طولانی تر (2-2.5 کیلوبایت) و قطعه الحاقی کوتاه (0.5-1.2 کیلوبایت) از ژل آگارز انتخاب می شوند. در حالی که قطعه های الحاقی بلند در ناقل های فاژ یا سوکمید کلون می شوند، قطعه های الحاقی کوتاه در ناقل های پلاسمید کلون می شوند. کتابخانه کلون قطعه الحاقی کوتاه برای توالی یابی از هر دو انتها استفاده می شود. از آنجایی که تعداد زیادی از کلون ها توالی یابی می شوند، هر یک از ژنوم ها بیش از 10 بار پوشش داده می شوند. از کلون های قطعه الحاقی طولانی می توان برای افزایش کارایی مونتاژ ژنوم استفاده کرد.
whole genome sequencing
مزایا
به نقشه های ژنومی نیاز ندارد.
زمان کمتر
صرفه جویی در پول
معایب
مونتاژ ژنوم برای ژنوم های یوکاریوتی به دلیل توالی های تکراری فراوان دشوار است.
توالی یابی ژنوم با استفاده از این روش دقیق نیست.
NGS در تسریع WGS نقش دارد.
بر خلاف رویکردهای کتابخانه ای مبتنی بر کلون، پلتفرم های توالی یابی نسل بعدی (NGS) از روش ساده سازی چشمگیری برای ساخت کتابخانه استفاده می کنند که توالی یابی کل ژنوم تفنگ ساچمه ای را ساده و تسریع کرده است. به طور کلی، DNA ژنومی ابتدا به طور تصادفی با استفاده از فراصوت یا نبولیزاسیون قطعه قطعه میشود و سپس به مجموعهای از آداپتورهای دو رشتهای مخصوص پلتفرم متصل میشود تا یک کتابخانه شاتگان ایجاد شود. متعاقبا، این قطعات کتابخانه را می توان در محل با هیبریداسیون و گسترش از آداپتورهای مکمل که به صورت کووالانسی به سطح یک سلول میکروسیال شیشه ای یا یک مهره کوچک (بسته به پلت فرم توالی) متصل شده اند، تقویت کرد. همه ابزارهای NGS از یک دستگاه میکروسیال برای حاوی قطعات تقویت شده کتابخانه شاتگان استفاده می کنند و به دنبال آن یک مرحله تصویربرداری که داده ها را از قطعاتی که به طور فعال توالی یابی می شوند جمع آوری می کند.
whole genome sequencing
ما توالیسنج Illumina را به عنوان مثال برای نشان دادن گردش کار WGS بر اساس توالییابی با توان بالا در نظر میگیریم.
ساخت کتابخانه توالی توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
ابتدا ژنوم آماده می شود و سپس DNA به طور تصادفی به صدها باز یا قطعه کوتاه تر با آداپتورهای خاص در هر دو انتها تقسیم می شود. اگر گروه رونویسی توالی داشته باشد، ساخت کتابخانه کمی مشکل تر است. پس از تکه تکه شدن RNA، باید به cDNA برگردد، سپس اتصال دهنده را اضافه کند، یا ابتدا RNA را به cDNA معکوس کند، سپس قطعه قطعه شده و اتصال دهنده را اضافه کند. اندازه قطعه (اندازه قطعه الحاقی) بر تجزیه و تحلیل داده های بعدی تأثیر دارد و می تواند بر اساس نیاز انتخاب شود. برای توالی یابی ژنوم، معمولاً چندین اندازهقطعه الحاقی مختلف انتخاب می شود تا هنگام مونتاژ اطلاعات بیشتری به دست آید.
- اتصال سطحی و تقویت پل
واکنش توالی یابی Solexa در یک لوله شیشه ای به نام فلوسل انجام می شود و فلوسل به 8 لاین تقسیم می شود که هر کدام دارای تعدادی اتصال تک رشته ای ثابت در سطح داخلی هر Lane هستند. قطعه DNA اتصالی به یک رشته تبدیل شده و با پرایمرهای کانال توالی یابی ترکیب میشود تا ساختاری مانند پل را برای مراحل بعدی تشکیل دهد.
- دناتوراسیون و تقویت کامل
dNTP بدون برچسب و آنزیم معمولی Taq برای تقویت PCR فاز جامد اضافه شده و نمونه پل تک رشته ای به یک قطعه پل دو رشته ای تکثیر میشوند. با دناتوره شدن، یک رشته تک رشته ای آزاد می شود و به سطح جامد مجاور متصل می شود. با چرخه مداوم، میلیون ها خوشه از آنالیت های دو رشته ای روی سطح جامد سلول فلو به دست می آید.
- گسترش تک جفت و توالی یابی
چهار dNTPs نشاندار شده با فلورسنت، DNA پلیمرازها و آغازگرهای پیوند دهنده برای تقویت به سلول های در جریان توالی یابی اضافه میشوند. هنگامی که هر خوشه توالی یابی رشته مکمل را گسترش میدهد، هر dNTP با برچسب فلورسنت اضافه می شود تا فلورسانس مربوطه را آزاد کند. توالیسنج اطلاعات توالی قطعه مورد آزمایش را با گرفتن یک سیگنال فلورسنت و تبدیل سیگنال نوری به پیک توالییابی توسط نرمافزار کامپیوتری به دست میآورد. طول خوانده شده تحت تأثیر تعدادی از عواملی است که باعث تضعیف سیگنال می شوند، مانند برش ناقص نشانگرهای فلورسنت. با افزایش طول خواندن، میزان خطا نیز افزایش می یابد.
- تحلیل داده ها
این مرحله صرفاً بخشی از فرآیند توالی نیست، اما فقط از طریق کار در مقابل این مرحله معنا پیدا میکند. دادههای خام بهدستآمده از توالییابی، دنبالهای به طول تنها چند ده باز است، و مجموعههایی که این توالیهای کوتاه را از طریق ابزارهای بیوانفورماتیک جمعآوری میکنند، چارچوب کل ژنوم هستند. متناوباً، این توالیها با یک ژنوم موجود یا توالی ژنوم گونهای مشابه همتراز میشوند و برای به دست آوردن نتایج بیولوژیکی معنیدار بیشتر مورد تجزیه و تحلیل قرار میگیرند.
کاربرد توالی یابی نسل سوم در توالی یابی کل ژنوم
اگرچه توالی یابی نسل بعدی، تجزیه و تحلیل در مقیاس جمعیتی انواع کوچک را ممکن کرده است، شناسایی تغییرات ساختاری بزرگتر دشوار است. علاوه بر این، مونتاژ de novo با استفاده از توالییابی نسل بعدی در مقایسه با مواردی که از روشهای قدیمیتر و گرانتر استفاده میکنند، اغلب از کیفیت پایینتری برخوردار است. فنآوریهای توالییابی تک مولکولی میتوانند بر این مشکلات فائق آیند که میتوانند تقریباً کل بازوهای کروموزوم را دربر گیرند و به محتوای GC حساس نیستند. فناوریهای توالییابی نسل سوم برای تولید مجموعههای جدید و مرجع بسیار دقیق برای میکروارگانیسمها، گیاهان، حیوانات و انسانها مورد استفاده قرار گرفتهاند که بینش جدیدی را در مورد تکامل و تنوع توالیها ممکن میسازد.
نمونه هایی از پلتفرم های توالی یابی نسل بعدی توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
چندین شرکت بر توسعه و بازاریابی ماشینهای توالییابی نسل بعدی (اغلب به عنوان پلتفرمها) برای استفاده در توالییابی کل ژنوم (و سایر موارد) تمرکز میکنند. Illumina به دلیل تعداد کاربرانی که از سیستم های آن استفاده می کنند، توسط بسیاری به عنوان پیشرو در نظر گرفته می شود. ایلومینا بسته به نیاز پلتفرم های متعددی دارد. Illumina HiSeq یکی از متداولترین ترتیبدهندههایی است که در آزمایشگاهها، از جمله مؤسسات تحقیقاتی بزرگ، شرکتهایی که خدمات توالییابی نسل بعدی را برای کلینیکها و آزمایشگاهها ارائه میکنند، و آزمایشگاههای پاتولوژی یافت میشود. توان عملیاتی بالایی دارد و قادر است ژنوم های زیادی را به سرعت با هزینه های معقول توالی یابی کند. این ابزار همچنین میتواند برای بررسی تغییرات تعداد نسخهها، جهشها و سایر تغییرات، و سطوح بیان RNA برای انجام رونویسی استفاده شود. به دلیل محبوبیت در کلینیک پانلهای توالییابی هدفمند، که بسیار کوچکتر هستند و کلینیکهایی که به زمانهای برگشت سریعتر برای درمان بیماران نیاز دارند، Illumina MiSeq را ایجاد کرد که میتواند نتایج توالییابی یک روزه را برای پانلهای بسیار کوچک ارائه دهد. Illumina همچنین تغییرات متعددی را برای ارائه ترتیبدهندهها برای هر ناحیه بیماری تولید کرد که خروجی، زمان چرخش و هزینهها را برای موارد استفاده خاص بهینه میکرد.
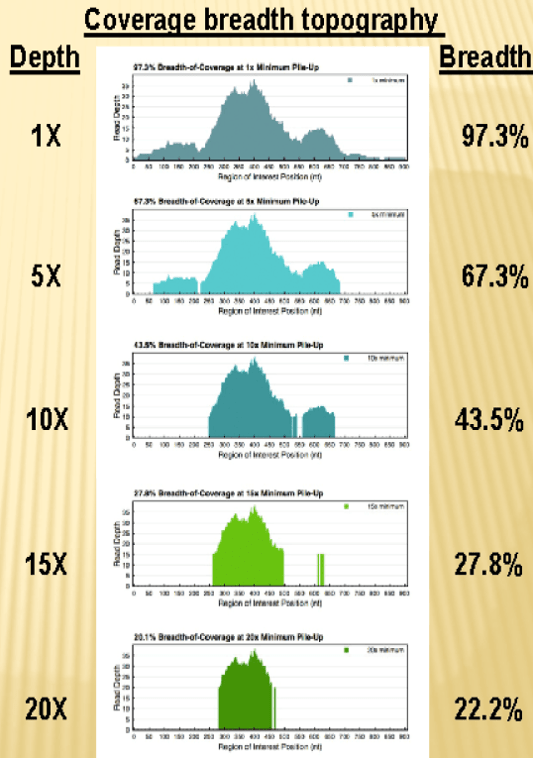
وسعت و عمق پوشش
پوشش به تعداد قرائت هایی اشاره دارد که یک نوکلئوتید خاص را در توالی DNA بازسازی شده نشان می دهد. خوانده شده رشته ای از بازهای A، T، C، G است که با DNA مرجع مطابقت دارد. میلیون ها مورد خوانده شده در یک اجرای توالی وجود دارد. افزایش عمق پوشش منجر به افزایش اطمینان در شناسایی انواع می شود.
برای ژنوم انسان، عمق پوشش 10 تا 30 برابر برای تشخیص جهش ها، SNP ها و بازآرایی ها قابل قبول است. رویکرد توالییابی نسل بعدی که عمق پوشش 30 برابری را فراهم میکند، پوشش بالایی در نظر گرفته میشود. با این حال، با افزایش عمق پوشش، وسعت پوشش کاهش می یابد.
whole genome sequencing
توالی یابی کل ژنوم در مقابل پانل های توالی یابی هدفمند ( توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing)
توالی یابی کل ژنوم، ترتیب نوکلئوتیدها (A، C، G، T) را در کل ژنوم تشکیل دهنده یک ارگانیسم تعیین می کند. هدف از توالی یابی کل ژنوم، به طور معمول، جستجوی انحرافات ژنتیکی است (به عنوان مثال، انواع تک نوکلئوتیدی، حذف ها، درج ها و انواع تعداد کپی). از آنجایی که کل ژنوم در حال تعیین توالی است، تغییرات در بخشهای غیرکدکننده DNA درون ژنها به نام اینترون نیز قابل تعیین است. در شرایط عادی، اینترون ها با پیوند RNA در طول یک فرآیند پس از رونویسی حذف می شوند، و تغییرات در این نواحی می تواند مهم باشد که آیا DNA به RNA رونویسی می شود یا به طور بالقوه منجر به یک پروتئین کوتاه و غیرعملکردی می شود.
یک رویکرد جایگزین، توالیبندی تنها اگزومها است که توالییابی کل اگزوم نامیده میشود. اگزوم ها بخشی از ژنوم هستند که توسط اگزون ها یا نواحی کد کننده تشکیل می شوند که وقتی رونویسی و ترجمه می شوند به پروتئین بیان می شوند. اگزوم ها تنها حدود 2 درصد از کل ژنوم را تشکیل می دهند. از آنجایی که ژنوم بسیار بزرگتر است، اگزوم ها می توانند در عمق بسیار بیشتر (تعداد دفعاتی که یک نوکلئوتید مشخص می شود) با هزینه کمتر توالی یابی شوند. این عمق بیشتر اطمینان بیشتری را در تغییرات فرکانس پایین فراهم می کند. عمق توالییابی میتواند حتی با هزینه کمتر با استفاده از یک پانل توالییابی هدفمند یا «نقطه داغ» که دارای تعداد معینی از ژنهای خاص یا مناطق کدگذاری درون ژنهایی است که دارای جهشهایی هستند که در پاتوژنز بیماری نقش دارند، بیشتر شود. ممکن است شامل ژن های مورد علاقه قابل عمل بالینی باشد (به عنوان مثال، تشخیصی، ترانوستیک، و غیره). اینها اغلب در مراقبت های بالینی برای ایجاد اطمینان بیشتر و همچنین کاهش هزینه و ایجاد فرصت بهتر برای بازپرداخت بیمه استفاده می شوند. با این حال، توالی یابی کل اگزوم و پانل های هدف تنها بخشی از داستان را می بینند زیرا بر روی مناطق کاهش یافته ژنوم تمرکز می کنند. در نتیجه، برای برخی پروژههای تحقیقاتی یا آزمایشهای ژنتیک، توالییابی کل ژنوم ممکن است سودمند باشد.
نقاط قوت و محدودیت های توالی یابی نسل بعدی در توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
نقاط قوت
نقطه قوت اصلی توالی یابی نسل بعدی این است که این روش می تواند ناهنجاری ها را در کل ژنوم تشخیص دهد (فقط توالی یابی کل ژنوم)، از جمله جایگزینی، حذف، درج، تکرار، تغییر تعداد کپی (ژن و اگزون) و وارونگی/جابه جایی کروموزوم. نقطه قوت اصلی توالییابی نسل بعدی این است که میتواند تمام آن ناهنجاریها را با استفاده از DNA کمتری نسبت به روشهای سنتی توالییابی DNA تشخیص دهد. توالی یابی نسل بعدی نیز هزینه کمتری دارد و زمان چرخش سریع تری دارد.
محدودیت ها
چندین محدودیت برای استفاده از توالی نسل بعدی وجود دارد. توالی یابی نسل بعدی اطلاعاتی در مورد تعدادی از انحرافات مولکولی ارائه می دهد. برای بسیاری از ناهنجاری های شناسایی شده، اهمیت بالینی در حال حاضر ناشناخته است. توالییابی نسل بعدی به سیستمهای بیوانفورماتیک پیچیده، پردازش سریع دادهها و قابلیتهای ذخیرهسازی بزرگ داده نیاز دارد که میتواند پرهزینه باشد. اگرچه بسیاری از موسسات ممکن است توانایی خرید تجهیزات توالی یابی نسل بعدی را داشته باشند، بسیاری از آنها فاقد منابع محاسباتی و کارکنان برای تجزیه و تحلیل و تفسیر بالینی داده ها هستند.
زمان و هزینه توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
زمان انجام اکثر روش های توالی یابی نسل بعدی و دریافت نتایج بسیار کاهش یافته است. از روزی که آزمایشگاه نمونه تومور را دریافت می کند، تقریباً 10 روز طول می کشد تا پزشک گزارش توالی یابی کل ژنوم را دریافت کند.
هزینه های توالی یابی کل ژنوم انسان در دهه گذشته به میزان قابل توجهی کاهش یافته است. در سال 2006، هزینه تقریباً 20 تا 25 میلیون دلار بود. در سال 2016، هزینه تعیین توالی ژنوم انسان به طور کلی کمتر از 1000 دلار است.
اهمیت بیوانفورماتیک
رشته علوم کامپیوتر به نام بیوانفورماتیک برای تجزیه و تحلیل داده های توالی یابی کل ژنوم استفاده می شود. این شامل الگوریتم، خط لوله و توسعه نرم افزار، و تجزیه و تحلیل، انتقال و ذخیره سازی/توسعه پایگاه داده داده های ژنومیک است.
یک گردش کار توالی یابی کل ژنوم معمولی شامل مراحل زیر است:
کنترل کیفیت و نظافت داده ها
مونتاژ ژنوم و/یا فراخوانی نوع
تجزیه و تحلیل پس از مونتاژ توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing
حجم داده ای که از پلتفرم های توالی یابی نسل بعدی تولید می شود، بسیار زیاد است. داده های جمع آوری شده نه تنها به نتایج توالی یابی DNA بلکه به عملکرد توالی یابی برای کمک به تشخیص خطاها یا توالی یابی مکرر مربوط می شود. این مشکلات مدیریت داده و ذخیره سازی را نشان می دهد. علاوه بر این، نرم افزار ویژه و سیستم های محاسباتی سریع برای پردازش داده های عظیم مورد نیاز است. بیوانفورماتیکان متخصص و آموزش دیده برای تجزیه و تحلیل داده های تولید شده توسط توالی یابی نسل بعدی و همچنین موفقیت و رشد مداوم پزشکی دقیق ضروری هستند
توالی یابی کامل ژنوم انسانی | Whole Genome Sequencing





علی –
نسبت به سایر مراکز بسیار با انصاف و دقیق
با تشکر از شما
graliontorile –
After all, what a great site and informative posts, I will upload inbound link – bookmark this web site? Regards, Reader.