تماس بگیرید
تماس بگیرید
تماس بگیرید
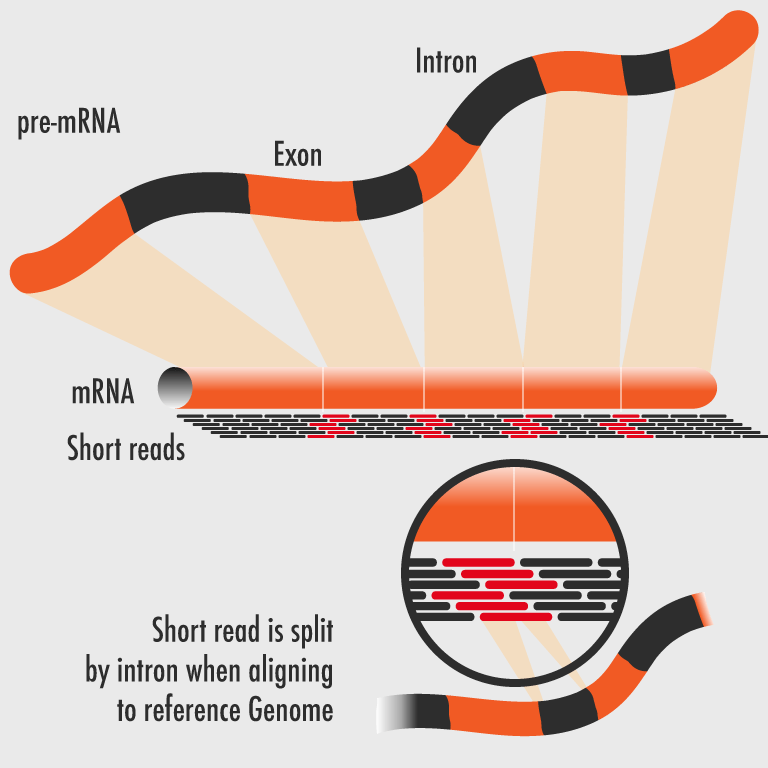


Hiseq X Ten, Whole Genome Sequencing
سیستم Hiseq X10 اولین سکانس کننده ی جهان است که سدهای سکانس کل ژنوم انسانی را شکسته است. این سیستم بازدهی بسیار بالا و قیمت نسبتا پایینی دارد. سیستم Hiseq مرزهای اقتصادی و مقیاسی توالی یابی کل ژنوم را در رابطه با گونه های انسانی و غیر انسانی جا به جا کرده است. این سیستم جهت توالی یابی کل ژنوم در مقیاس جمعیت نیز مناسب است. این نوع سکانس می تواند مسیر را جهت تهیه ی منبعی جامع از تنوع های (variation) انسانی و غیر انسانی، تهیه ی رفرنس های مبتنی بر جمعیت، دستیابی به اکتشافات دور از ذهن و تسهیل درک عمیق از زیست شناسی و بیماری های ژنتیکی هموار کند.
سری HiSeq X از سیستم های توالی یابی (sequencing)
حداکثر توان و کمترین هزینه برای توالی یابی کل ژنوم در مقیاس جمعیت
شرکت Illumina به دنبال نوآوری مستمر در توالی یابی ژنوم انسان با افزایش توان داده ها در هر سال به بیش از دوبرابر موانع را از بین برده است، این در حالیست که به طور چشمگیری قیمت کاهش یافته است. از طرف دیگر این شرکت برای اولین بار توالی یابی ژنوم را در پوشش 30× در طول یک روز برای سلول های سالم و یا سرطانی انجام داده و اکنون فناوری Illumina به محققان کمک می کند تا به نقطه عطف دیگری برسند و توسط دستگاه HiSeq X Ten فقط با1000 دلار ژنوم انسان را توالی یابی نمایند. در نتیجه می توان امیدوار بود با توان عملیاتی فوق العاده بالا HiSeq X Ten و قیمت پایین بی سابقه برای توالی یابی کل ژنوم هر نمونه، رویای توالی یابی در مقیاس جمعیت (WGS) به واقعیت تبدیل شود.)جدول1 (سیستم HiSeq X Five نیز با نیاز به سطح پایینتری از سرمایهگذاری اولیه، Whole Genome Sequencing دقیق و قابل دسترس را برای هزاران نمونه در سال با قیمت کمی بالاتر و در عین حال مقرون به صرفه برای هر ژنوم انسان ارائه میکند. آزمایشگاههایی که با HiSeq X Five شروع می کنند و سپس ظرفیت خود را به 10 ابزار یا بیشتر افزایش میدهند، میتوانند عملکرد HiSeq X Ten و توالی یابی ژنوم انسان با قیمت 1000 دلاری را ممکن کنند.)جدول 1(
|
HiSeq X Ten |
HiSeq X Five |
|
|
حداقل تعداد دستگاه |
10 |
5 |
|
توان سالانه (تعداد ژنوم) |
> 18,000 |
> 9000 |
|
قیمت با ازای توالی یابی 30x |
< $1000 |
< $1500 |
گسترش تکنیک در گونه های غیر انسانی
سیستم های HiSeq X اکنون می توانند توالی یابی در مقیاس جمعیت را برای گونه های غیر انسانی ممکن کنند. این سیستم پوشش بالایی در زمینه های مختلف، از جمله کشاورزی و تحقیقات ارگانیسم های مدل در صنعت داروسازی ارائه می دهد و با ارائه توان عملیاتی بسیار بالا و قیمت بیسابقهای برای هر ژنوم، فرصتی را برای مشتریان فراهم میکند تا اقتصاد و مقیاس توالییابی کل ژنوم را فراتر از نوع انسان تغییر دهند.

سیستم HiSeq X Ten مجموعه ای از 10 ابزار جداگانه HiSeq X است.
فناوری نوآورانه، عملکرد اثبات شده
سیستمهای HiSeq X Ten و HiSeq X Five از توالییابی تایید شده Illumina توسط روش سنتز (SBS) استفاده میکنند، که بهطور گسترده در فناوری نسل جدید توالییابی مورد استفاده قرار میگیرد. این روش کیفیت داده ها را تضمین می کند و به محققان اطمینان کامل را در کسب نتایج خوب می دهد (جدول 2). سری HiSeq X با تکیه بر این روش قدرتمند، شرکت ایلومینا از فناوری جریان سلول بر اساس الگو(Patterned flow cell technology) برای تولید توان عملیاتی عظیم استفاده می کند. در این روش میلیاردها نانو چاه در مکانهای مشخص ثابت هستند، خوشهبندی اختصاصی صورت می پذیرد و تکثیر حذفی، تضمین میکند که تنها یک الگوی DNA به یک چاه متصل شده و یک خوشه را تشکیل میدهد و در نتیجه حداکثر خروجی داده را به همراه خواهد داشت.
بهترین روش آماده سازی کتابخانه
برای دستیابی به پوشش استثنایی ژنوم برای تشخیص دقیق و جامع تغییرات ژنومی، HiSeq X Ten و HiSeq X Five از 2 کیت آماده سازی کتابخانه پشتیبانی می کنند. کیت آماده سازی کتابخانه PCR-Free TruSeq، که یک پروتکل سریع و بدون ژل برای آماده سازی کتابخانه های WGS با پوشش عالی مناطقی که به طور سنتی توالی یابی آنها دشوار است، مانند مناطق غنی از GC، پروموترها و محتوای تکراری، ارائه می دهد. آمادهسازی بدون PCR، سوگیری و شکافهای کتابخانه را کاهش میدهد، و در نتیجه کیفیت دادهای بینظیر برای شناسایی بیشترین تعداد انواع مختلف ایجاد میکند. کیت آماده سازی کتابخانه TruSeq Nano DNA Library امکان توالی یابی کارآمد نمونه ها را با کمتر از 100 نانوگرم DNA فراهم را می کند. گردش کار افزایش یافته تعداد و اندازه متوسط شکاف های معمولی ناشی از PCR در پوشش را کاهش می دهد، سوگیری کتابخانه را به حداقل می رساند و یکنواختی پوشش را در سراسر ژنوم بهبود می بخشد و با استفاده از پروتکل ساده TruSeq Nano DNA، کتابخانه ها را می توان در کمتر از 1 روز آماده کرد.
|
پارامتر |
مشخصات |
|
خروجی در هر سلول |
جریان دوگانه: 1.6-1.8 ترابایت |
|
Single Reads Passing Filter |
جریان دوگانه: 5.3-6 میلیارد |
|
پشتیبانی از طول خواندن |
2 × 150 جفت بر ثانیه |
|
زمان اجرا |
< 3 روز |
|
کیفیت |
≥ 75٪ از پایه های بالاتر از Q30 در 2 × 150 جفت باز |
|
کیت آماده سازی کتابخانه |
TruSeq DNA PCR-Free Library Prep Kit TruSeq Nano DNA Library Prep Kit |
مشخصات مبتنی بر کتابخانه کنترل Illumina PhiX در تراکم خوشه های پشتیبانی شده (1255–1412 K clusters/mm2) در 1 سیستم HiSeq X می باشد.
شکل 2: سلولهای جریان الگو حاوی میلیاردها نانو چاه در مکانهای ثابت هستند که فاصله خوشهای یکنواخت و اندازه ویژگی یکنواخت را برای ارائه چگالی بسیار بالای خوشه فراهم میکنند.
LIMS کاملاً یکپارچه
Illumina یک روش کار کاملاً خودکار برای سری HiSeq X ارائه می دهد که شامل BaseSpace® Clarity LIMS X Edition، رباتیک حمل مایعات Hamilton Microlab STAR و تجهیزات کمکی تعریف شده برای ارائه ردیابی کامل نمونه مثبت است. این سیستم از پیش پیکربندی شده است تا از هر دو جریان کاری TruSeq DNA PCR-Free و TruSeq Nano DNA با استفاده از Illumina Automated Workflow Manager for Hamilton پشتیبانی کند و رابط کاربری بصری امکان پذیرش سریع سیستم را برای ردیابی فوری فرآیند و مقیاس پذیری فراهم می کند.
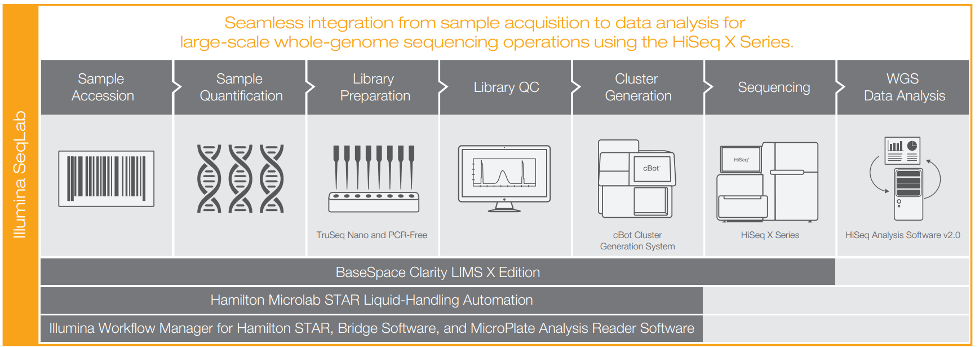
شکل 3: Illumina SeqLab Workflow ادغام یکپارچه از پیوستن نمونه به تجزیه و تحلیل داده ها با استفاده از روش ها و نرم افزار
خلاصه
سیستمهای HiSeq X Ten و HiSeq X Five همچنان به شکستن موانع توالییابی ادامه میدهند، اقتصاد توالییابی کل ژنوم را تغییر میدهند و پایهای خواهند بود را برای دانشمندان، مؤسسات و کشورهایی که برای انجام تحقیقات پیشگامانه تلاش میکنند و برای همیشه درک ما را از ژنوم انسان بالا می برند. درست زمانی که پروژههای توالییابی ژنوم در مقیاس جمعیت در سراسر جهان شروع به شکلگیری میکنند، سری HiSeq X ژنومهای انسانی و غیرانسانی واقعاً مقرون به صرفه را در مقیاس عظیم ارائه میدهد. سری HiSeq X با ایجاد ظرفیت برای توالی یابی هزاران تا ده ها هزار ژنوم، توالی یابی ژنوم را به نقطه عطف می رساند. این سیستمهای با کارایی بالا راه را برای فهرستی جامع از تنوع انسانی و غیرانسانی، اکتشافات گسترده و تسریع درک عمیقتر از بیولوژی و بیماریهای ژنتیکی هموار خواهند کرد.
Whole Exome Sequencing
با کمک توالی یابی نسل بعدی، اکنون می توان مقادیر زیادی از DNA را توالی یابی کرد، به عنوان مثال تمام قطعات DNA یک فرد که دستورالعمل هایی برای ساخت پروتئین ها ارائه می دهند. تصور می شود که این قطعات که اگزون نامیده می شوند، 1 درصد از ژنوم یک فرد را تشکیل می دهند. همه اگزون های یک ژنوم با هم به عنوان اگزوم شناخته می شوند و روش تعیین توالی آنها با عنوان توالی یابی کل اگزوم شناخته می شود. این روش اجازه می دهد تا تغییرات در ناحیه کدکننده پروتئین همه ی ژن ها شناسایی شود، نه فقط در چند ژن منتخب. از آنجایی که بیشتر جهش های شناخته شده ای که باعث بیماری های ژنتیکی می شوند در اگزون ها شناسایی شده اند، تصور می شود که توالی یابی کل اگزوم یک روش کارآمد برای شناسایی جهش های احتمالی ایجاد کننده بیماری ها باشد.
با این حال، محققان دریافتهاند که تغییرات DNA خارج از اگزونها میتواند بر فعالیت ژن و تولید پروتئین تأثیر بگذارد و منجر به اختلالات ژنتیکی شود – تغییراتی که توالییابی کل اگزومها قادر به تشخیص آن ها نیست. توالییابی کل ژنوم، ترتیب تمام نوکلئوتیدها را در DNA یک فرد تعیین میکند و میتواند تغییرات را در هر بخشی از ژنوم تعیین کند.
این روش هزینه بسیار کمتری در مقایسه با توالی یابی کل ژنوم دارد. واریانت های اگزومی می توانند مسئول ایجاد بیماری های مندلی و یا پلی ژنیک همچون آلزایمر باشند. توالی یابی اگزوم می تواند به عنوان روشی در تحقیقات و همچنین در تشخیص های کلینیکی استفاده شود.
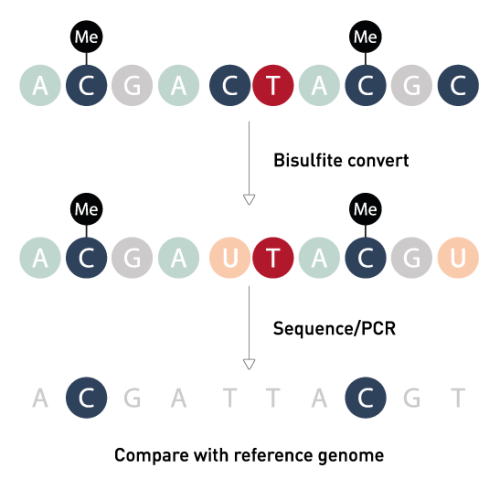
Human Whole Genome Bisulfite Sequencing
توالی یابی بی سولفیت کل ژنوم یک فناوری توالی یابی نسل بعدی است که برای تعیین وضعیت متیلاسیون DNA با سیتوزین های منفرد با تیمار DNA با بی سولفیت سدیم قبل از تعیین توالی DNA ، استفاده می شود. وضعیت متیلاسیون DNA در ژنهای مختلف میتواند اطلاعات مربوط به تنظیم بیان ژن و فعالیتهای رونویسی را نشان دهد.
توالی یابی بی سولفیت کل ژنوم سطوح متیلاسیون تک سیتوزینی را در کل ژنوم اندازه گیری می کند و مستقیماً نسبت مولکول های متیله شده به سطوح غنی سازی را تخمین می زند. در حال حاضر، این تکنیک تقریباً 95 درصد از تمام سیتوزین ها را در ژنوم های شناخته شده شناسایی کرده است. با بهبود روش های آماده سازی کتابخانه و فناوری توالی یابی نسل بعدی در دهه گذشته، توالی یابی بی سولفیت کل ژنوم به روشی گسترده برای تجزیه و تحلیل متیلاسیون DNA در مطالعات گسترده اپی ژنومیک تبدیل شده است.
RNA-sequencing
يكي از كاربردهاي مهم و در حال گسترش NGS ، توالي يابي RNAكل است. با استفاده از اين تكنيك امكان بررسي تمام انواع مختلف نسخه هاي موجود در سلول اعم از RNA هاي كد كننده و غيركد كننده (ترانسكريپتوم) وجود دارد. تكنيك RNA-seq براي انجام مطالعات ژنتيكي و مولكولي به ويژه در موجودات غير مدل كه اطلاعات ژنتيكي بسيار اندكي از آنها موجود است بسيار حائز اهميت و پر كاربرد مي باشد. با استفاده از اين روش تصوير تقريبا كاملي از ترانسكريپتوم، از يك نمونه بيولوژيك حاصل مي شود كه اطلاعات قابل استخراج از آن در برخي موارد تا حد زيادي مي تواند جايگزين توالي يابي كل ژنوم گردد، اين در حالي است كه متد سنگر تنها قادر به شناسايي ٦٠% از نسخه هاي موجود در سلول است. به منظور توالي يابيRNA ، پس از تبديل اين مولكول به cDNA عمدتا از تكنولوژي هاي توالي يابي 454 و Illumina استفاده مي شود، به علت هزينه پايين تر و مقدار بيشتر توالي قرائت شده توسط Illumina ، اين تكنولوژي به ويژه در سال هاي اخير مورد توجه محققان مختلف قرار گرفته است. تعيين ساختار نسخه برداري ژن ها، مطالعه ي الگوهاي پردازش RNA و سایر تغييرات بعد از نسخه برداري، شناسايي چند شكلي هاي تك نوكلئوتيدي SNP و همچنين آناليز كمي بيان ژن ها در مقياس وسيع از كاربردهاي مهم RNA-seq مي باشد، امروزه اين تكنيك جايگزين ميكرواري براي بررسی بيان ژن ها در مقياس وسيع شده است. پس از توالي يابي RNA با اين متد، در صورتي كه ژنوم موجود مورد نظر تعيين توالي شده باشد، توالي هاي خوانده شده با توالي هاي ژنومي هم رديف شده و سپس آناليز مي شوند در غير اينصورت ابتدا توالي هاي خوانده شده براي ايجاد يك ژنوم ساختگي اسمبل مي شوند و سپس توالي هاي قرائت شده با اين ژنوم هم رديف مي گردند.
لازم به ذكر است كه اسمبلي ترانسكريپتوم و آناليز توالي هاي حاصل شده در طی RNA-seq به ويژه در موجوداتي با حداقل اطلاعات ژنتيكي بزرگترين چالش در اين حوزه مي باشد و موفقيت در آن مستلزم دانش و تخصص كافي و استفاده از برنامه ها و نرم افزارهاي كامپيوتري پيشرفته است. با اين وجود، از آنجايي كه شناسايي ژن هاي جديد و يا ايزوفرم هاي جديد ژن هاي شناخته شده و همچنين توالي يابي بخش هايي از ژنوم كه به ميزان بالايي متحمل بازآرايي شده اند (مانند آنچه در ژنوم سلول هاي سرطانی وجود دارد) مستلزم De novo assembly است، كميت و كيفيت برنامه ها و نرم افزارهاي مورد نياز دائما در حال افزايش و بهبود است.
Nova Seq
توالی یابی کل ژنوم را کارآمدتر و مقرون به صرفه تر از همیشه انجام می دهد. NovaSeqسریع است، اما برای کاربران آزمایشگاه ، کاهش زمان اجرا در مقایسه با HiSeq 4000 تأثیر چشمگیری بر کل زمان نجام پروژههای معمولی RNA-Seq یا Exome ندارد. پروژه های بزرگتر تنها 40 ساعت سریعتر به پایان می رسند.
ChIP-Seq (HiSeq2500 SE50)
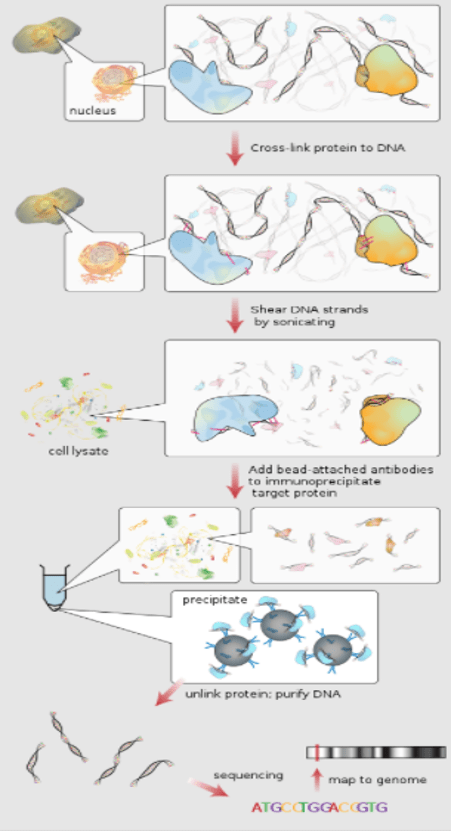
ChIP-Sequencing که با نام ChIP-Seq نیز شناخته می شود، روشی برای تجزیه و تحلیل برهمکنش های پروتئین با DNA می باشد. ChIP-seq رسوب ایمنی کروماتین (chromatin immunoprecipitation) را با توالی های DNA های موازی انبوه جهت شناسایی محل اتصال پروتئین های مرتبط با DNA ترکیب می کند. از این تکنیک می توان برای تهیه ی نقشه مکان های اتصال هر پروتئینی استفاده کرد. پیش از این، ChIP-on-chip رایجترین تکنیکی بود که برای مطالعه ارتباط پروتئین و DNA استفاده میشد.
ChIP-seqدر ابتدا جهت تعیین چگونگی اثر فاکتورهای رونویسی و سایر پروتئین های مرتبط با کروماتین بر مکانیسم های موثر در شکل گیری فنوتیپ استفاده می شد. تعیین نحوه تعامل پروتئین ها با DNA برای تنظیم بیان ژن، جهت درک کامل بسیاری از فرآیندهای بیولوژیکی و بیماری ها ضروری می باشد.
مکانهای DNA خاص که در تعامل فیزیکی مستقیم با فاکتورهای رونویسی و سایر پروتئینها هستند را میتوان با رسوب ایمنی کروماتین جدا کرد. این تکنیک، کتابخانه ای از سایت های DNA متصل به پروتئین مورد نظر را ایجاد می کند. تجزیه و تحلیل توالی موازی این اطلاعات انبوه در ترکیب با داده های حاصل از توالی یابی کل ژنوم جهت تجزیه و تحلیل الگوی تعامل هر پروتئین با DNA، یا الگوی هر گونه تغییرات کروماتین اپی ژنتیکی استفاده میشود.
از جمله مطالعات انجام شده با استفاده از اين تكنيك، مي توان به تعيين موقعيت نوكلئوزوم ها در سطح DNA با استفاده از تكنولوژي توالي يابي ٤٥٤ و تعيين جايگاه اتصال برخي از مهمترين فاكتورهاي نسخه برداري به DNA با استفاده از سيستم توالي يابي Illumina اشاره كرد.
مراحل انجام تکنیک ChIP-seq را می توانید به صورت خلاصه در تصویر زیر مشاهده نمایید.
Metatranscriptome sequencing
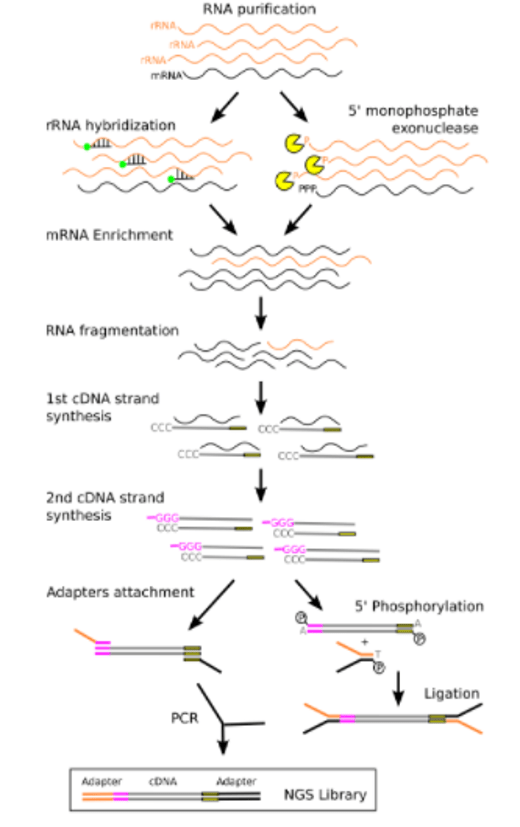
توالییابی متاترانسکریپتوم به مطالعه عملکرد بیان ژن ها در نمونه های محیطی، در یک زمان خاص اشاره دارد. این تکنیک که در واقع به وسیله RNA-Sequencing انجام می شود، اطلاعاتی در رابطه با ژن های با میزان بیان بالا در یک محیط میکروبی خاص به ما می دهد.
توالییابی متاترانسکریپتوم، نیاز به جداسازی و کشت باکتری ها ندارد. این تکنیک بیان ژن ها را با توالییابی تصادفی mRNAها ارائه میکند و پویایی الگوهای بیان ژن در جوامع میکروبی را در طول زمان با استفاده از توالییابی نسل جدید (NGS) ردیابی میکند. این مسئله درک ما را از ساختار، عملکرد و مکانیسمهای تطبیقی جوامع پیچیده بهبود میبخشد.
کاربردهای مهم توالییابی متاترانسکریپتوم:
- شناسایی باکتریهای فعال عملکردی و تعامل متابولیک آن ها در تحقیقات اکولوژی میکروبی.
- شناسایی پاسخ ایمنی و تحریک فرآیندهای خودایمنی در تحقیقات بالینی.
- کمک به کشف منابع جدید دارو رسانی که امروزه به دلیل محدودیت در جداسازی میکروبی به راحتی قابل دسترسی نیستند.
- بهبود کیفیت غذا و دادههای مرتبط با رفتارهای متابولیکی جوامع میکروبی.
- شناسایی ژن های جدید و تشخیص تغییرات در جوامع میکروبی پیچیده در سطح رونویسی.
مراحل انجام تکنیک Metatranscriptome sequencing را می توانید به صورت خلاصه در تصویر زیر مشاهده نمایید.
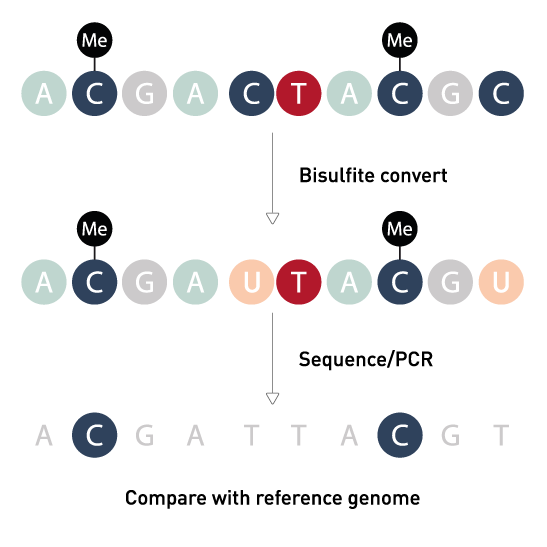
Human Targeted Methylation Sequencing
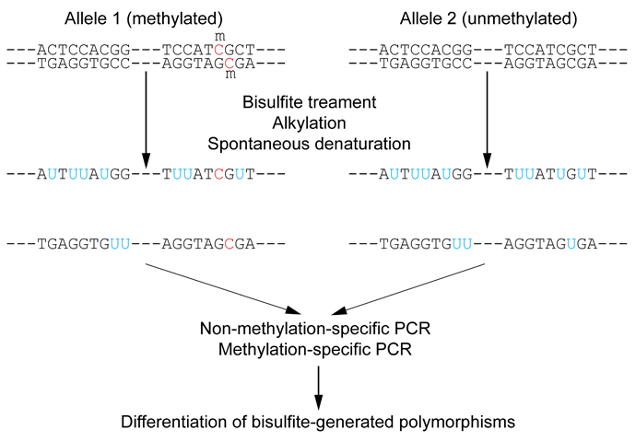
متیلاسیون DNA تغییر اپی ژنتیکی مهمی است که بر چندین فرآیند رشد حیاتی تأثیر می گذارد. متیلاسیون سیتوزین می تواند به طور قابل توجهی بیان ژن را به صورت موقت و یا دائمی و همچنین وضعیت کروماتین را تغییر دهد. توالی یابی متیلاسیون (Methyl-seq or bisulfite sequencing) ابزاری قدرتمند جهت درک متیلاسیون در سطح ژنوم با وضوح تک نوکلئوتیدی است و یک استاندارد طلایی محسوب می شود. در حال حاضر، توالییابی DNA متیله در یک سلول با استفاده از انواع روشهای تجربی امکانپذیر است و درک بیشتری را در مورد ارتباط بین فنوتیپ و ژنوتیپ سلول ارائه میکند. توالی یابی متیلاسیون برپایه ی تبدیل هر سیتوزین متیله نشده به اوراسیل متکی است، در واقع پرایمرهای اختصاصی برای منطقه مورد نظر طراحی شده و بازهای تبدیل شده (بعد از PCR) به عنوان تیمین در داده های توالی یابی شناسایی می شوند و از شمارش خوانده شده برای تعیین درصد سیتوزین های متیله استفاده می شود. تغییرات متیلاسیون سیتوزین در آن منطقه ارزیابی می شود. در صورتی که این تبدیل به صورت ناقص صورت گرفته باشد، در آنالیز داده ها، به اشتباه سیتوزین های متیله نشده ای که هنوز به اوراسیل تغییر نیافته اند، به عنوان سیتوزین های متیله تفسیر می شود، که منجر به نتایج مثبت کاذب برای متیلاسیون می شود.
متیلاسیون DNA بیان ژن را با به کارگیری پروتئین های دخیل در سرکوب ژن یا با مهار اتصال فاکتور(های) رونویسی به DNA تنظیم می کند. الگوی متیلاسیون DNA در ژنوم در نتیجه یک فرآیند پویا که هم متیلاسیون و هم دی متیلاسیون DNA را شامل می شود، تغییر می کند.
مزایای توالی یابی متیلاسیون:
- کشف الگوهای متیلاسیون نواحی CpG، CHH، و CHG را در سراسر ژنوم انسان
- مشاهده متیلاسیون در سیتوزین های سراسر ژنوم در اکثر گونهها با توالییابی بی سولفیت کل ژنوم (WGBS)
- کشف تنوع کامل ژنوم با استفاده از مقادیر بسیار کم DNA
- شناسایی مناطق نوظهور در ژنوم انسان توسط ENCODE، FANTOM5، و کنسرسیوم Epigenomics RoadMap با استفاده از توالی متیلاسیون هدفمند
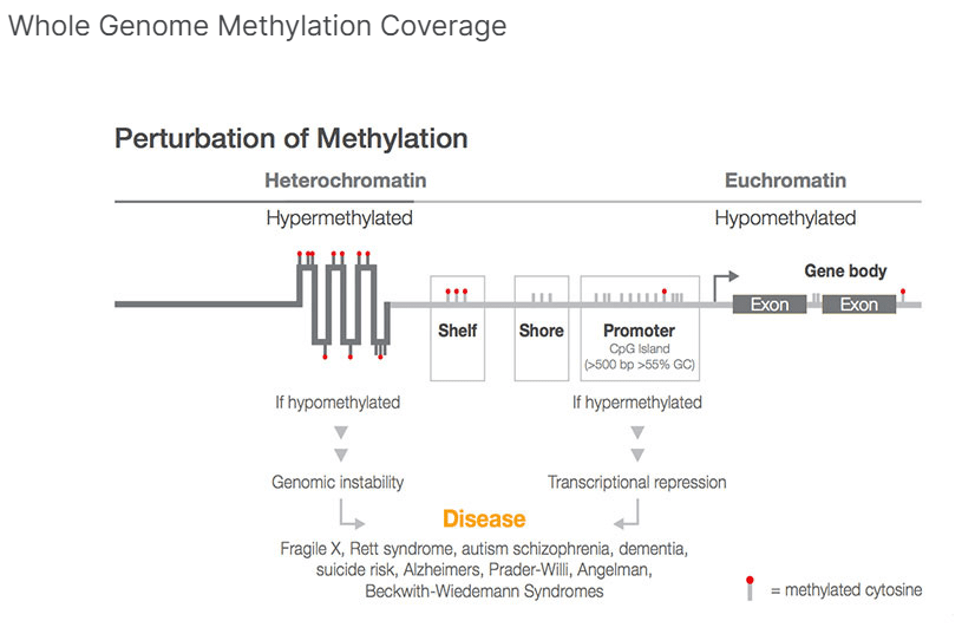
Genotyping-by-sequencing (GBS)
در زمینه توالی یابی ژنتیکی، تعیین ژنوتیپ از طریق توالی یابی که GBS نیز نامیده می شود، روشی برای کشف پلی مورفیسم های تک نوکلئوتیدی (SNP) به منظور انجام مطالعات ژنوتایپینگ، مانند مطالعات ارتباط گسترده ژنوم (GWAS) است. GBS از آنزیم های محدود کننده به منظور کاهش پیچیدگی ژنوم و تعیین ژنوتیپ چند نمونه DNA استفاده می کند. در واقع پس از هضم آنزیمی، PCR انجام میشود و سپس کتابخانههای GBS با استفاده از فناوریهای توالییابی نسل جدید توالییابی میشوند، که معمولاً منجر به خوانش حدود 100bp میشود. این تکنیک نسبتاً ارزان است و تا به حال به مقدار زیادی در اصلاح نباتات استفاده شده است.
GBS یک روش قوی، ساده و مقرون به صرفه جهت کشف و نقشه برداری SNP است. به طور کلی، این رویکرد پیچیدگی ژنوم را با آنزیمهای محدودکننده در گونههای ژنومی با تنوع بالا کاهش میدهد تا توالییابی با کارایی بالا انجام شود. با استفاده از آنزیم های محدود کننده های مناسب، می توان از نواحی تکراری ژنوم جلوگیری کرد و نواحی با کپی پایین تر را هدف قرار داد، که مشکلات هم ترازی را در گونه های ژنتیکی بسیار متنوع کاهش می دهد. این روش برای اولین بار توسط الشر و همکارانش توصیف شد. به طور خلاصه، DNAهای با وزن مولکولی بالا با استفاده از یک آنزیم محدودکننده خاص که قبلاً با برش مکرر در بخش عمده تکراری ژنوم تعریف شده بود، استخراج و هضم میشوند. ApeKI پر استفاده ترین این آنزیم ها در این تکنیک است. سپس آداپتورهای بارکد به انتهای چسبنده متصل می شوند و PCR انجام می شود. فناوری توالی یابی نسل جدید انجام می شود که منجر به خوانش حدود 100 جفت باز می شود. دادههای توالی خام با استفاده از ابزار همترازی Burrows-Wheeler (BWA) یا Bowtie 2 فیلتر شده و با ژنوم مرجع تراز میشوند. هنگامی که یک SNP در مقیاس بزرگ و در سطح گونه یافت شد، می توان به سرعت SNP های شناخته شده را در نمونه های جدید توالی یابی کرد.
Chloroplast Sequencing
کلروپلاست اندامکی درون سلولهای گیاهان و جلبکهای خاص است که محل فتوسنتز در آن هاست، که طی آن انرژی خورشید برای رشد به انرژی شیمیایی تبدیل میشود. توالی یابی گیاهی اولین گام به سوی درک زیربنای ژنتیکی صفات و تعاملات یک موجود زنده با محیط است. روشی جامع برای تجزیه و تحلیل کل ژنوم زمانی که ژنوم مرجع یک گونه موجود باشد. میتوکندری ها و کلروپلاست ها در یک فرآیند هماهنگ رشد می کنند که به دو سیستم ژنتیکی جداگانه نیاز دارد، یکی در اندامک و دیگری در هسته سلول. بیشتر پروتئین های موجود در این اندامک ها توسط DNA هسته ای کدگذاری می شوند، در سیتوزول سنتز می شوند و سپس به صورت جداگانه وارد این اندامک ها می شوند. پلاستید و میتوکندری اندامک های ضروری در سلول های گیاهی هستند. مطالعه ی ژنوم کلروپلاست (cp) و ژنوم میتوکندری (mt) اغلب برای مطالعه تکامل گیاهان استفاده می شود. اطلاعات تمام ژنومهای cp توالییابی شده نشان داده که طول اکثر آنها بین 120 تا 160 کیلوبایت است و دارای محتوای GC 30 تا 40 درصد می باشند
Proteomics
پروتئومیکس مطالعه پروتئین ها در مقیاس بالاست. پروتئین ها بخش های حیاتی موجودات زنده هستند که عملکردهای زیادی دارند. پروتئوم کل مجموعه ای از پروتئین های تولید شده یا اصلاح شده توسط یک ارگانیسم یا سیستم است. تکنیک پروتئومیکس امکان شناسایی تعداد روزافزون پروتئین ها را فراهم می کند. پروتئومیکس یک حوزه بین رشته ای است که از اطلاعات ژنتیکی پروژه های مختلف ژنوم، از جمله پروژه ژنوم انسانی، بهره زیادی برده است. این اکتشاف تمامی سطوح ترکیب، ساختار و فعالیت پروتئین ها را پوشش می دهد و جزء مهمی از ژنومیک عملکردی است.
اولین مطالعات پروتئین هایی که می توانند به عنوان پروتئومیکس در نظر گرفته شوند، در سال 1975، پس از معرفی ژل دو بعدی و نقشه برداری از پروتئین های باکتری اشریشیا کلی آغاز شد.
پروتئوم ترکیبی از دو کلمه “پروتئین” و “ژنوم” است که در سال 1994 توسط مارک ویلکینز دانشجوی دکترا در دانشگاه مک کواری، که اولین آزمایشگاه اختصاصی پروتئومیکس را در سال 1995 تأسیس کرد، ابداع شد.
پس از ژنومیکس و ترانسکریپتومیکس، پروتئومیکس گام بعدی در مطالعه سیستم های بیولوژیکی می باشد که پیچیده تر از ژنومیکس است زیرا ژنوم یک موجود زنده کم و بیش ثابت است، در حالی که پروتئوم ها از سلولی به سلول دیگر و از زمان به زمان متفاوت است. در انواع مختلف سلول ژن های متمایزی بیان می شوند، به این معنی که حتی مجموعه اصلی پروتئین های تولید شده در یک سلول نیز باید شناسایی شوند.
در گذشته این پدیده با تجزیه و تحلیل RNA مورد ارزیابی قرار می گرفت، که مشخص شد با محتوای پروتئین همبستگی ندارد. اکنون مشخص شده است که mRNA همیشه به پروتئین ترجمه نمی شود و مقدار پروتئین تولید شده برای مقدار معینی از mRNA به ژنی که از آن رونویسی می شود و به وضعیت فیزیولوژیکی سلول بستگی دارد. پروتئومیکس وجود پروتئین را تأیید می کند و اندازه گیری مستقیم مقدار آن را ارائه می دهد.
در پروتئومیکس، روش های متعددی برای مطالعه پروتئین ها وجود دارد. به طور کلی، پروتئین ها ممکن است با استفاده از آنتی بادی ها (ایمونواسی) یا طیف سنجی جرمی شناسایی شوند. اگر یک نمونه بیولوژیکی پیچیده تجزیه و تحلیل شود، یا باید از یک آنتی بادی بسیار خاص در تجزیه و تحلیل کمی نقطهای (QDB) استفاده شود، یا جداسازی بیوشیمیایی باید قبل از مرحله تشخیص استفاده شود.
تشخیص پروتئین با آنتی بادی ها (ایمونواسی):
آنتیبادیهای پروتئینهای خاص یا اشکال اصلاحشده آنها در مطالعات بیوشیمی و زیستشناسی سلولی مورد استفاده قرار گرفتهاند. آنتی بادی ها یکی از رایج ترین ابزارهایی هستند که امروزه توسط زیست شناسان مولکولی استفاده می شود. چندین تکنیک و پروتکل خاص وجود دارد که از آنتی بادی ها برای تشخیص پروتئین استفاده می کنند. سنجش ایمونوسوربنت متصل به آنزیم (ELISA) برای دههها جهت شناسایی و اندازهگیری کمی پروتئینها در نمونهها استفاده شده است. وسترن بلات به منظور تشخیص و تعیین کمیت پروتئین های منفرد استفاده می شود، جایی که در مرحله اولیه، یک مخلوط پروتئین پیچیده با استفاده از SDS-PAGE جدا می شود و سپس پروتئین مورد نظر با استفاده از یک آنتی بادی شناسایی می شود. پروتئین های تغییریافته را می توان با ایجاد یک آنتی بادی خاص برای آن تغییر مورد مطالعه قرار داد. به عنوان مثال، آنتیبادیهایی وجود دارند که پروتئینهای خاصی را فقط زمانی تشخیص میدهند که تیروزین در آن ها فسفریله شوند، آنها به عنوان آنتیبادیهای اختصاصی فسفو شناخته میشوند. همچنین، آنتیبادیهایی خاص برای سایر تغییرات وجود دارد. اینها ممکن است برای تعیین مجموعه ای از پروتئین هایی که دستخوش تغییرات مورد نظر شده اند استفاده شوند.
تشخیص پروتئین بدون آنتی بادی:
در حالی که تشخیص پروتئین با آنتیبادیها هنوز در زیستشناسی مولکولی بسیار رایج است، روشهای دیگری نیز توسعه یافتهاند که به آنتیبادی متکی نیستند. این روش ها مزایای مختلفی را ارائه می دهند، به عنوان مثال اغلب قادر به تعیین توالی یک پروتئین یا پپتید هستند، ممکن است توان عملیاتی بالاتری نسبت به آنتی بادی داشته باشند، و گاهی اوقات می توانند پروتئین هایی را که هیچ آنتی بادی برای آنها وجود ندارد شناسایی و تعیین کنند. یکی از اولین روشها برای تجزیه و تحلیل پروتئین، تجزیه ادمن (معرفی شده در سال 1967) است که در آن یک پپتید منفرد تحت چندین مرحله تجزیه شیمیایی قرار میگیرد تا توالی آن را حل کند. این روشهای اولیه عمدتاً توسط فناوریهایی که توان عملیاتی بالاتری ارائه میدهند، جایگزین شدهاند. روشهایی که اخیراً اجرا شده اند از تکنیکهای مبتنی بر طیفسنجی جرمی استفاده میکنند، پیشرفتی که با کشف روشهای «یونیزاسیون نرم» مانند دفع/یونیزاسیون لیزری به کمک ماتریس (MALDI) و یونیزاسیون الکترواسپری (ESI) که در دهه 1980 توسعه یافت.
برای تجزیه و تحلیل نمونه های بیولوژیکی پیچیده، کاهش پیچیدگی نمونه مورد نیاز است. این ممکن است خارج از خط با جداسازی یک بعدی یا دو بعدی انجام شود. اخیراً، روشهای آنلاین توسعه یافتهاند که در آن پپتیدهای منفرد با استفاده از کروماتوگرافی فاز معکوس جدا میشوند و سپس مستقیماً با استفاده از ESI یونیزه میشوند.
طیف سنجی جرمی و پروفیل پروتئین:
در حال حاضر دو روش مبتنی بر طیف سنجی جرمی برای پروفایل پروتئین استفاده می شود. روش تثبیتشده و گستردهتر از الکتروفورز دو بعدی با وضوح بالا برای جدا کردن پروتئینها از نمونههای مختلف بهطور موازی، و به دنبال آن انتخاب و رنگآمیزی پروتئینهای بیان شده متفاوت برای شناسایی توسط طیفسنجی جرمی استفاده میکند. روش کمی دوم از برچسب های ایزوتوپ پایدار برای برچسب گذاری متفاوت پروتئین ها از دو مخلوط پیچیده مختلف استفاده می کند. در این روش، پروتئینهای درون یک مخلوط پیچیده ابتدا به صورت ایزوتوپی برچسبگذاری میشوند و سپس برای تولید پپتیدهای نشاندار هضم میشوند. سپس مخلوطهای نشاندار شده با هم ترکیب میشوند، پپتیدها با کروماتوگرافی مایع چند بعدی جدا شده و با طیفسنجی جرمی پشت سر هم تجزیه و تحلیل میشوند. در این روش، باقیماندههای سیستئین پروتئینها به صورت کووالانسی به معرف ICAT متصل میشوند و در نتیجه پیچیدگی مخلوطها را کاهش میدهند و باقیماندههای غیر سیستئینی را حذف میکنند.
Protein chips:
جهت متعادل کردن استفاده از طیفسنجهای جرمی در پروتئومیکس و در پزشکی، از ریزآرایههای پروتئینی استفاده می شود. هدف اصلی ریز آرایه های پروتئینی چاپ هزاران ویژگی تشخیص پروتئین جهت شناسایی نمونه های بیولوژیکی است. Antibody arrays ها مجموعهای از آنتیبادیهای مختلف برای شناسایی آنتیژنهای مربوطه خود از نمونه خون انسان هستند. رویکرد دیگر، آرایهبندی انواع پروتئینهای متعدد برای مطالعه خواصی مانند برهمکنشهای پروتئین-DNA، پروتئین-پروتئین و پروتئین-لیگاند است. در حالت ایدهآل، آرایههای پروتئومی عملکردی شامل کل مکمل پروتئینهای یک موجود زنده میشوند. اولین نسخه از این آرایه ها شامل 5000 پروتئین خالص شده از مخمر بود که بر روی اسلایدهای میکروسکوپی شیشه ای قرار گرفتند. علیرغم موفقیت اولین تراشه، اجرای آرایه های پروتئینی چالش بزرگتری بود. کار با پروتئین ها بسیار دشوارتر از DNA است. آنها محدوده دینامیکی وسیعی دارند، از پایداری کمتری نسبت به DNA برخوردارند و حفظ ساختار آنها بر روی اسلایدهای شیشه ای دشوار است، اگرچه برای اکثر سنجش ها ضروری هستند. فناوری جهانی ICAT دارای مزایای قابل توجهی نسبت به فناوری های تراشه پروتئینی است.
ریزآرایه های پروتئین فاز معکوس:
این تکنیک یک برنامه ریزآرایه امیدوارکننده و جدیدتر برای تشخیص، مطالعه و درمان بیماری های پیچیده مانند سرطان است. این فناوری، ریزشکن کردن لیزر (LCM) را با فناوری میکرو آرایه ادغام میکند تا ریزآرایههای پروتئین فاز معکوس تولید کند. در این نوع ریزآرایهها، کل مجموعه پروتئین خود با هدف ثبت مراحل مختلف بیماری در یک بیمار بیحرکت میشوند. هنگامی که از LCM استفاده می شود، آرایه های فاز معکوس می توانند وضعیت نوسان پروتئوم را در میان جمعیت سلولی مختلف در ناحیه کوچکی از بافت انسانی نظارت کنند که برای تعیین وضعیت مولکولهای سیگنالدهنده سلولی، در میان مقطعی از بافت که شامل سلولهای طبیعی و سرطانی است، مفید است. این رویکرد در نظارت بر وضعیت عوامل کلیدی در اپیتلیوم طبیعی پروستات و بافت های سرطانی مهاجم پروستات مفید می باشد. سپس LCM این بافتها را تشریح میکند و لیزهای پروتئینی روی لامهای نیتروسلولزی قرار می گیرند و با آنتیبادیهای خاص بررسی می شوند. این روش میتواند انواع رویدادهای مولکولی را ردیابی کند و میتواند بافتهای بیمار و سالم را در یک بیمار مقایسه کند و امکان توسعه استراتژیهای درمانی و تشخیص را فراهم کند. توانایی به دست آوردن عکس های فوری پروتئومیکس از جمعیت های سلولی همسایه، با استفاده از ریزآرایه های فاز معکوس در ارتباط با LCM، تعدادی کاربرد فراتر از مطالعه تومورها دارد. این رویکرد می تواند بینش هایی در مورد فیزیولوژی و آسیب شناسی طبیعی همه بافت ها ارائه دهد و برای توصیف فرآیندهای رشد و ناهنجاری ها بسیار ارزشمند است.
از مهم ترین کاربردهای پروتئومیکس می توان به موارد زیر اشاره کرد:
- کشف داروهای جدید
- برهمکنش پروتئین ها و شبکه های پروتئینی
- شنلسایی پروتئین های بیانی
- مارکرهای زیستی




