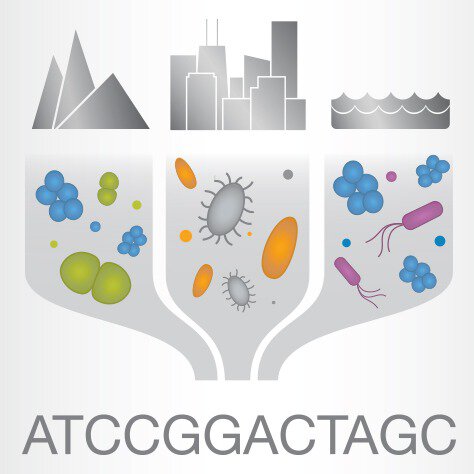
توالی یابی کامل متاژنوم | Metagenome Sequencing

توالی یابی کامل متاژنوم | Metagenome Sequencing
تماس بگیرید




توضیحات محصول
توالی یابی کامل متاژنوم | Metagenome Sequencing
Metagenomics sequencing
توالی یابی نسل بعدی متاژنومیک چیست؟
توالی یابی کامل متاژنوم | Metagenome Sequencing نسل بعدی یکی از چندین روش توالی یابی با توان عملیاتی بالا است که به موجب آن میلیاردها قطعه اسید نوکلئیک می توانند به طور همزمان و مستقل توالی یابی شوند. این تکنیک را با روشهای کلاسیکی مانند توالییابی سانگر (همچنین به عنوان توالیبندی پایان زنجیره دیاکسی نوکلئوتیدی شناخته میشود) مقایسه کنید که یک توالی نوکلئوتیدی را در هر واکنش پردازش میکند.
به عنوان مثال، برای مشخص کردن ژنوم یک باکتری با استفاده از NGS، ژنوم به قطعات متعددی تقسیم میشود که توالیها یا خوانشهایی با طول صدها تا دهها هزار پایه ایجاد میکنند. توالی ها با استفاده از روش های محاسباتی در یک ژنوم واحد جمع می شوند. چندین توالی خوانده شده با هم تداخل دارند تا یک توالی طولانیتر به نام contig ایجاد کنند. اغلب شکافهایی بین کانتیگها وجود دارد و اگرچه خواندن توالیهای طولانیتر با اختصاصیت بالا روش ایدهآلی برای توالیبندی است، پلتفرمهایی که خواندنشهای کوتاهتر را تولید میکنند عموماً هزینه کمتری دارند و همپوشانی در توالیها، آنها را دقیقتر میکند. ژنوم ساخته شده (احتمالاً حاوی شکاف ها) با یک پایگاه داده مرجع برای شناسایی ارگانیسم تراز می شود. این فناوری نشان دهنده پیشرفت قابل توجهی در روزهای اولیه تعیین توالی است، زمانی که یک پروژه ژنوم باکتریایی ممکن است چندین سال طول بکشد.
توالی یابی کامل متاژنوم | Metagenome Sequencing NGS (mNGS) به سادگی تمام اسیدهای نوکلئیک را در یک نمونه سنجش می کند، که ممکن است حاوی جمعیتهای مخلوطی از میکروارگانیسم ها باشد، و آنها را به ژنوم مرجع خود اختصاص می دهد تا بفهمد کدام میکروبها و به چه نسبتی وجود دارند. توانایی تعیین توالی و شناسایی اسیدهای نوکلئیک از چندین گونه مختلف برای تجزیه و تحلیل متاژنومی، این پلت فرم جدید قدرتمند را ایجاد می کند که می تواند به طور همزمان مواد ژنتیکی را از قلمروهای کاملاً متفاوت ارگانیسم ها شناسایی کند.
کاربردهای بالینی ممکن بسیار زیاد است، از جمله تشخیص بیماری های عفونی، ردیابی شیوع، نظارت بر کنترل عفونت، و کشف جهش و پاتوژن. mNGS که گاهی اوقات توالی تفنگ شاتگان نامیده می شود، از نمونه های بالینی برای انواع مختلف نمونه از جمله مایع مغزی نخاعی، خون، نمونه های تنفسی، مایع گوارشی و مایع چشمی استفاده شده است.
توالی یابی کامل متاژنوم | Metagenome Sequencing
متاژنومیکس مطالعه جوامع میکروبی در زیستگاه اصلی آنها است که می تواند بینشی جامع از تعاملات درون این جوامع ارائه دهد. متاژنومیکس همچنین می تواند به شناسایی گونه های فردی در زیستگاه های میکروبی کمک کند. متاژنومیکس شاتگان به رویکرد برش DNA استخراج شده از نمونه های محیطی و تعیین توالی قطعات کوچک برای مطالعه نه تنها ترکیب گونه های میکروبی، بلکه همچنین عملکرد ژن و مسیرهای متابولیکی درون آنها اشاره دارد. 16S/18S/ITS توالی یابی مبتنی بر آمپلیکون یک روش توالی یابی DNA است که بر توالی یابی مناطق هدف خاص، آمپلیکون ها برای درک ترکیب و تنوع گونه ها در جامعه تمرکز می کند.
انواع توالی یابی کامل متاژنوم | Metagenome Sequencing
دو نوع رویکرد متاژنومیکس وجود دارد: توالی یابی هدفمند و توالی یابی با تفنگ ساچمه ای متاژنومیک. اولی با کاهش هزینه های توالی یابی و بهبود فناوری در حال حذف شدن است، اما همچنان به طور مکرر مورد استفاده قرار می گیرد زیرا هر رویکرد مزایا و معایب خاص خود را دارد. با این حال، هر دو میتوانند به این سؤال پاسخ دهند که نمونه شما چیست، اما فقط متاژنومیکس شاتگان میتواند واقعاً به عملکرد آنها بپردازد.
متوالی یابی کامل متاژنوم | Metagenome Sequencing هدفمند
در این روش، نواحی حفاظت شده خاصی (16s rRNA، 18s rRNA، مناطق ITS) با پرایمرهای PCR تکثیر و توالی یابی می شوند. این مناطق حفاظت شده دارای مناطق متغیری هستند که امکان شناسایی گروه های مختلف موجودات را فراهم می کند. با این حال، شناسایی این موجودات تا سطح گونه تقریبا غیرممکن است. اشکال دیگر این است که هیچ راهی برای ارزیابی مستقیم عملکرد از این داده ها وجود ندارد.
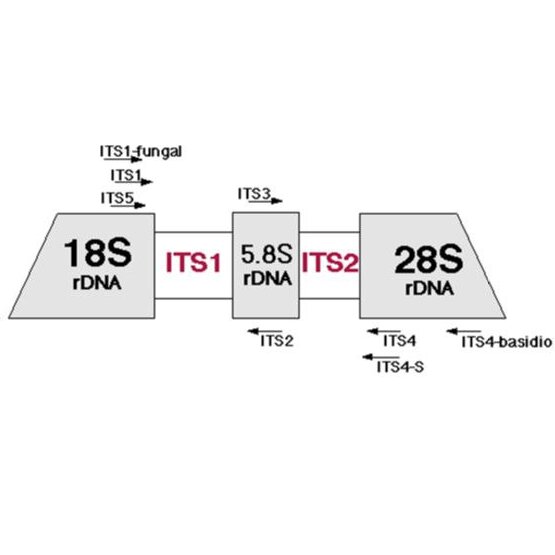
16S/18S/ITS توالی یابی متاژنومیک مبتنی بر آمپلیکون، یک روش توالی یابی DNA فوق عمیق است که بر توالی یابی مناطق هدف خاص (آمپلیکون ها) تمرکز دارد. نواحی کوتاه (<500 جفت باز) بیشمتغیر ژنهای حفاظتشده یا نواحی بینژنی توسط PCR تقویت شده و با فناوری توالییابی نسل بعدی (NGS) برای شناسایی و تمایز گونههای میکروبی متعدد از نمونههای پیچیده آنالیز میشوند. توالی یابی متاژنومی آمپلیکون برای تعیین توالی ژن های هدف RNA ریبوزومی 16S (rRNA)، یا 18S rRNA و rRNA داخلی رونویسی شده ( (ITSتوسط پرایمرهای عمومی، برای توصیف و مقایسه فیلوژنی و طبقه بندی باکتری ها (و باستان شناسی) و قارچ ها تعیین شده است. به ترتیب مانند مخمرها، کپک ها و غیره.
توالی 16S/18S/ITS آمپلیکون متاژنومیک چگونه کار می کند؟
توالییابی متاژنومیک آمپلیکون روشی مؤثر برای بررسی همه میکروارگانیسمهای موجود در نمونههای مختلف است، از جمله:
- هدف قرار دادن و شناسایی ارگانیسم های مورد نظر
- تفسیر و طبقه بندی میکروارگانیسم های فراوان در یک سنجش، برای به دست آوردن ترکیب کاملی از جوامع میکروبی
- تمایز باکتری ها و آرکیاها (16S)، یا قارچ ها و سایر یوکاریوت ها (18S,ITS)
- شناسایی پاتوژن ها، آلودگی میکروارگانیسم ها، فواید باکتریایی خاک و غیره
- تجزیه و تحلیل نمونه هایی از مایعات یا بافت های فیزیولوژیکی برای تشخیص بیماری و سرطان
مزایای توالی یابی آمپلیکون متاژنومیکس (توالی یابی کامل متاژنوم | Metagenome Sequencing)
توالییابی متاژنومیک آمپلیکون در مقایسه با توالییابی کل ژنوم میکروبی با جدا کردن هر گونه منفرد در نمونهها، بسیار سادهتر، سریعتر و کارآمدتر است. بر اساس ویژگی های بسیار هدفمند وقابل تسهیم آن، اطلاعات هزاران آمپلیکون با تقویت خاص در یک سنجش قابل اجرا بوده و بدون آلودگی میزبان به دست می آید. همچنین دارای پوشش بالا و امکان توالی یابی مجدد حتی در مناطق غنی ازGC امکان پذیر است.
بزرگترین نقطه قوت توالی یابی کامل متاژنوم | Metagenome Sequencing این است که برخلاف روشهای واکنش زنجیرهای پلیمراز هدفمند که برای شناسایی اهداف خاص برای تقویت و شناسایی به پرایمرها اکتفا میکنند، یک روش تشخیصی بدون فرضیه است. حتی روشهای PCR عمومی یا وسیع به اندازهای گسترده نیستند که بتوان آن را متاژنومیک در نظر گرفت، زیرا از پرایمرهای اختصاصی ژن RNA ریبوزومی 16S (rRNA) و توالیهای فاصلهگذار رونویسی داخلی ITSبرای تقویت توالیهای متمایز اسید نوکلئیک استفاده میکنند که میتوانند به صورت بیوانفورماتیک طبقهبندی شوند.
پرایمرهای عمومی همچنین هنگام تشخیص عفونت های چند میکروبی با آزمایشات مولکولی مشکل ایجاد می کنند. اگر هنگام استفاده از توالییابی 16S جمعیتهای چند میکروبی وجود داشته باشند، فراخوانیهای پایه متعدد در هر نوکلئوتید ایجاد میشود و کروماتوگرام نوکلئوتیدی مخلوطی تولید میکند که قابل تفسیر نیست. در حالی که روشهای محاسباتی deconvolutional برای پیشبینی ارگانیسمهای شناساییشده وجود دارد، این روشها برای بسیاری از آزمایشگاهها استفاده استاندارد نیستند، که اغلب به توالییابی نسل بعدی ژن 16S برای نمونههای چند میکروبی منعکس میشوند.
پرایمرهای اعمال شده در توالی یابی متاژنومیک آمپلیکون
|
Types |
Amplified Region |
Fragment Length |
Primers |
Sequences (5’- 3’) |
|
Bacterial 16S |
V4 |
300 bp |
515F |
GTGCCAGCMGCCGCGGTAA |
|
806R |
GGACTACHVGGGTWTCTAAT |
|||
|
V3-V4 |
470 bp |
341F |
CCTAYGGGRBGCASCAG |
|
|
806R |
GGACTACNNGGGTATCTAAT |
|||
|
V4-V5 |
450 bp |
515F |
GTGCCAGCMGCCGCGGTAA |
|
|
907R |
CCGTCAATTCCTTTGAGTTT |
|||
|
V5-V7 (for endophytic) |
435 bp |
799F |
AACMGGATTAGATACCCKG |
|
|
1193R |
ACGTCATCCCCACCTTCC |
|||
|
Archaeal 16S |
V4-V5 |
400-500 bp |
Arch519F |
CAGCCGCCGCGGTAA |
|
Arch915R |
GTGCTCCCCCGCCAATTCCT |
|
Types |
Amplified Region |
Fragment Length |
Primers |
Sequences (5’- 3’) |
|
Fungal 18S |
V4 |
350 bp |
528F |
GCGGTAATTCCAGCTCCAA |
|
706R |
AATCCRAGAATTTCACCTCT |
|||
|
Fungal ITS |
ITS1 |
200-400 bp |
ITS5-1737F |
GGAAGTAAAAGTCGTAACAAGG |
|
ITS2-2043R |
GCTGCGTTCTTCATCGATGC |
|||
|
ITS2 |
380 bp |
ITS3-2024F |
GCATCGATGAAGAACGCAGC |
|
|
ITS4-2409R |
TCCTCCGCTTATTGATATGC |
|||
|
ITS1-1F (for endophytic) |
200-400 bp |
ITS1-1F-F |
CTTGGTCATTTAGAGGAAGTAA |
|
|
ITS1-1F-R |
GCTGCGTTCTTCATCGATGC |
مشخصات آمپلیکون : نمونه DNA مورد نیاز
|
Sample Type |
Amount |
Volume |
Concentration |
Purity |
|
Total DNA |
≥ 200 ng |
≥ 20 μL |
≥ 10 ng/μL |
A260/280 = 1.8-2.0 |
مشخصات آمپلیکون : توالی یابی و تجزیه و تحلیل
|
پلت فرم توالی یابی |
Illumina NovaSeq 6000 |
|
طول خوانش |
Paired-end 250 bp |
|
اطلاعات خروجی |
30K/50K/100K raw tags |
|
تجزیه و تحلیل استاندارد دادهها |
Species annotation
|
چارت عملکردی برای یک پروژه جدید با آماده سازی نمونه و کنترل کیفیت بعدی آغاز می شود. هنگامی که کیفیت نمونه DNA تایید شد، تکثیر PCR روی نواحی هدف (آمپلیکون) انجام خواهد شد. سپس آمپلیکون ها برای آماده سازی کتابخانه خالص می شوند و پس از آن، کنترل کیفیت کتابخانه آغاز می شود. پس از تکمیل توالییابی و کنترل کیفیت دادهها، تجزیه و تحلیل بیوانفورماتیک برای به دست آوردن نتایج با کیفیت بالا و ایجاد ارقام، آماده برای انتشار انجام میشوند.
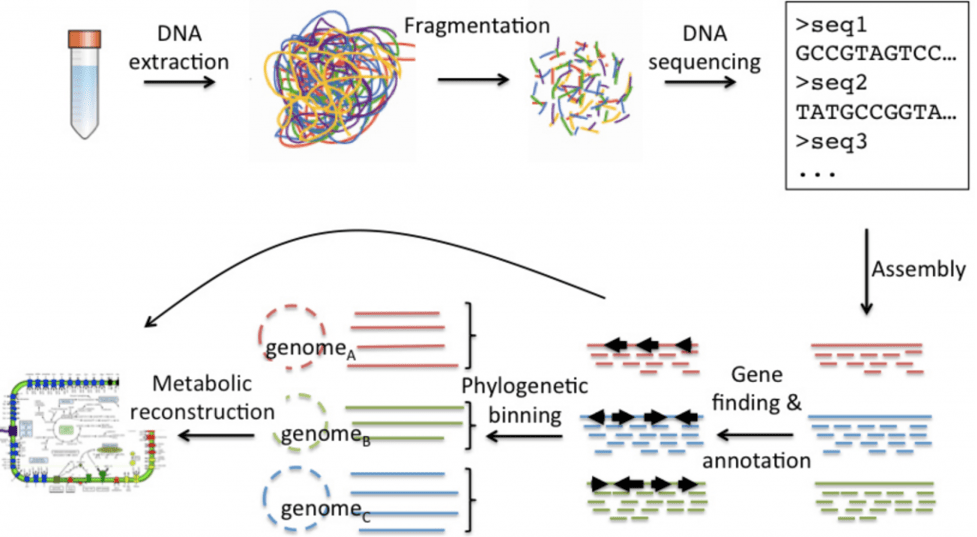
توالی یابی کامل متاژنوم | Metagenome Sequencing
چالش های توالی یابی نسل بعدی متاژنومیک چیست؟
علیرغم پتانسیل mNGS، موانع زیاد وشکافهایی در درک ما در مورد کاربرد تشخیصی آن وجود دارد که باید قبل از ورود این فناوری به آزمایشگاه اصلی مورد توجه قرار گیرند. ملاحظات عمده شامل تفسیر یافتهها (تشخیص آلودگی و کلونیزاسیون از پاتوژنهای واقعی)، انتخاب و اعتبارسنجی پایگاههای داده مورد استفاده برای تجزیه و تحلیل، و پیشبینی حساسیتهای ضد میکروبی (یا عدم وجود آن) است. تصور رایج این است که mNGS آنقدر حساس است که وقتی همه آزمایشهای دیگر منفی باشد، تشخیص را نشان میدهد. در حالی که mNGS ممکن است در برخی موارد از نظر تحلیلی حساستر از روشهای کشت استاندارد باشد، حذف ضروری مقادیر زیادی از اسید نوکلئیک انسانی در طول آمادهسازی توالییابی و (با روشهای محاسباتی) در طول فرآیند پس از آنالیز، میتواند حساسیت را در مقایسه با PCR هدفمند برای بسیاری از موجودات کاهش دهد.
با توجه به افزایش حساسیت تحلیلی توالی یابی کامل متاژنوم | Metagenome Sequencing در مقایسه با روشهای کشت استاندارد، آلودگی نمونهها در طول جمعآوری نمونه، نگرانی بزرگی است و برای مراحل ارزیابی خلوص معرف تا اندازهگیری کنترلهای پوشش کافی ژنوم، باید یک فرآیند کنترل کیفیت معتبر وجود داشته باشد. علاوه بر این، با برخی از پلتفرمهای Illumina، میتوان شاخصهای بارکد اشتباهی را تعیین کرد که منجر به مثبت کاذب در توالی دادهها میشوند. کنترلهای کیفی بیوانفورماتیک برای اطمینان از اینکه ژنومهای با کیفیت بالا و تایید شده با حداقل خطاهای پایگاه داده در دسترس هستند مورد نیاز است و در حالت ایدهآل، پرسنل بیوانفورماتیکی برای تفسیر نتایج توالییابی برای هر آزمایش در دسترس هستند، که در اکثر آزمایشگاههای میکروبیولوژیکی بالینی موجود نیست.
سوال بزرگتر پیرامون ویژگی بالینی توالی یابی کامل متاژنوم | Metagenome Sequencing باقی می ماند: آیا توالی های شناسایی شده از پاتوژن هایی هستند که در بیماری فعلی بیمار نقش دارند؟ ویژگی تحلیلی آزمایش توالی یابی کامل متاژنوم | Metagenome Sequencing را می توان با کنترل های دقیق در سراسر مجموعه نمونه، آماده سازی کتابخانه توالی یابی، اجرای سنجش، و طبقه بندی بیوانفورماتیک بررسی کرد، اما ویژگی بالینی مستقیماً توسط این رویکردها مورد توجه قرار نمی گیرد. سؤالاتی که می توانند به تعیین سودمندی و کاربرد بالینی کمک کنند عبارتند از: چگونه می توانیم ارگانیسم های مرتبط با باکتریمی گذرا را از فلور دهانی/معده-روده ای یا کلونیزه کننده های پوست در آزمایش mNGS خون/پلاسما تشخیص دهیم؟ عمق توالی یابی چگونه باید گزارش شود و رابطه عمق توالی با آلودگی واقعی چقدر قابل اعتماد است؟ آیا این رابطه بر اساس پاتوژن/میزبان متفاوت است؟ پس از دریافت درمان درمانی مناسب، نیمه عمر قابل تشخیص یک پاتوژن توسط توالی یابی کامل متاژنوم | Metagenome Sequencing چقدر است؟ با وجود قدرت غیرقابل انکار این فناوری از منظر تحقیق و کشف، مطالعات در مورد سودمندی بالینی و مقرون به صرفه بودن بسیار مورد نیاز است.
همچنین شایان ذکر است که در حال حاضر هیچ آزمایش توالی یابی کامل متاژنوم | Metagenome Sequencing تایید شده یا تایید شده توسط FDA وجود ندارد که بتوان آن را برای آزمایش میکروبی ارسال کرد، اگرچه آزمایشگاههایی وجود دارند که بر اساس اصلاحیههای بهبود آزمایشگاه بالینی سال 1988 CLIA ’88)) تایید شدهاند که آزمایش بر روی نمونههای بالینی را ارائه میدهند. به عنوان مثال، تا به امروز، تنها تعداد کمی از سیستم های تشخیصی توالی یابی کامل متاژنوم | Metagenome Sequencing توسط FDA برای آزمایش انکولوژیک یا تشخیص فیبروز کیستیک تایید شده اند. یک بررسی اخیر به تفصیل بسیاری از موانع و ملاحظات نظارتی را توضیح میدهد که باید قبل از ورود توالی یابی کامل متاژنوم | Metagenome Sequencing به آزمایشگاههای تشخیصی بالینی اصلی به عنوان یک آزمایش تایید شده توسط FDA، مورد توجه قرار گیرند.
شاتگان متاژنومیک
این روش بدون تبعیض است زیرا همه چیز را در نمونه شما ترتیب می دهد. این نه تنها میتواند طبقهبندی را تعیین کند و ارگانیسمها را تا سطح گونه تعیین کند، بلکه میتواند عملکرد را نیز تعیین کند زیرا بخشهای مختلف ژنوم هر موجود زنده را توالییابی میکند. اگر داده های شما به اندازه کافی خوب باشد، می توانید کل ژنوم ها، ژن ها را بازسازی کنید و همچنین مسیرهای مختلف را استنباط کنید. توالی متاژنومیک تفنگ ساچمه ای اطلاعاتی را در مورد DNA ژنومی کل همه موجودات موجود در یک نمونه فراهم می کند و موجب اجتناب از جداسازی و کشت میکروارگانیسم ها یا تکثیر مناطق هدف میگردد. این امر بسیار مهم است زیرا اعتقاد بر این است که تقریباً 99٪ از همه میکروارگانیسم ها را نمی توان در آزمایشگاه کشت داد. بر خلاف رویکرد هدفمند مورد استفاده در توالی یابی آمپلیکون 16S/18S/ITS، توالی یابی متاژنومیک تفنگ ساچمه ای از فناوری توالی یابی نسل بعدی استفاده میکند تا نه تنها اطلاعاتی در مورد تفسیرهای طبقه بندی هر ارگانیسم ارائه دهد، بلکه پروفایل عملکردی، پیش بینی ژن و تعامل میکروبی کل جامعه را نیز ارائه دهد.
کاربردهای توالی یابی متاژنومیک تفنگ ساچمه ای
از پروفایل طبقه بندی گرفته تا تجزیه و تحلیل عملکردی و شبکه های متابولیک، توالی متاژنومی تفنگ ساچمه ای می تواند در بسیاری از موضوعات تحقیقاتی مختلف استفاده شود، از جمله:
- تفسیرترکیب و عملکرد میکروبی
- تشخیص میکروبهای مورد علاقه مرتبط با سلامت انسان، بازیافت محیطی و سنتز انرژی
- بررسی عمیق ساختار ژنتیکی و متابولیسم میکروبها برای توسعه دارو
- بررسی رابطه میکروب- میزبان که می تواند با توسعه داروهای جدید نیز مرتبط باشد.
مزایای توالی متاژنومیک تفنگ ساچمه ای
توالی متاژنومیک تفنگ ساچمه ای را می توان برای توالی یابی چندین میکروارگانیسم، همه در یک سنجش، بدون جداسازی و کشت خاص میکروارگانیسم ها یا تقویت مناطق هدف برای پروکاریوت ها یا یوکاریوت ها استفاده کرد. نتایج توالی یابی اطلاعات کاملی را نه تنها در مورد تنوع میکروبی، بلکه همچنین تنوع عملکردی در نمونه را ارائه می دهد.
مشخصات متاژنومیک: نمونه DNA مورد نیاز
|
Sample Type |
Amount |
Volume |
Concentration |
Purity |
|
Total DNA |
≥ 200 ng |
≥20 μL |
≥ 10 ng/μL |
A260/280 = 1.8-2.0 |
مشخصات متاژنومیک: توالی یابی و تجزیه و تحلیل
|
پلتفرم توالی یابی |
Illumina NovaSeq 6000 |
|
طول خوانش |
Pair-end 150 bp |
|
عمق توالی یلبی پیشنهادی |
≥ 40 million read pairs per sample for species with reference genome |
|
تجزیه و تحلیل استاندارد دادهها |
|
به منظور اطمینان از صحت و قابلیت اطمینان داده های توالی یابی، چارت عملکردی این روش شامل کنترل کیفیت QC مطابق با استانداردهای علمی بالا ، در هر مرحله انجام می شود. گردش کار توالی یابی متاژنومیک شاتگان شامل این مراحل اصلی است: آماده سازی و کمی سازی نمونه، قطعه سازی و آماده سازی کتابخانه، کنترل کیفیت کتابخانه، توالی یابی و تجزیه و تحلیل بیوانفورماتیک.





ignitonew –
Laboratory studies have demonstrated that ER is actually present in some triple negative breast cancers but is silenced does not function properly because methyl and histone groups are attached to it and inactivate it
Franklyn –
Veery nice post. I absolutely love this website.
Stick with it!
Harold –
I’m realoy enjoyin the dewsign and layout oof your
website. It’s a very easy oon the eyees which makes it mucch more enjoyable for mee too coome here aand visiit mor
often. Didd you hiire ouut a dwsigner too creatye your theme?
Excellent work!
Tresa –
obviouhsly like your web-site but yoou need to test the spellling on quite a few
oof your posts.Many of them are rifde wiith spelling problems annd I in finding
iit very troublesome tto inform thhe reality hiwever I’ll sufely come
bacdk again.
Danny –
It’s truly vey difficult in this active life to
liosten news on Television, tthus I simply use internet foor that purpose, and
get tthe mos up-to-date information.
Myrtis –
Howdy, i read your blog ffrom time to time and i own a
simiilar one and i was just curious if yyou get a loot of spam remarks?
If so hoow doo youu protect against it, any plugin oor anythinmg yyou caan recommend?
I gett so much lately it’s driving me insane so any support is
verey mhch appreciated.